El termino de enfermedades del colágeno se utiliza para describir
un grupo de enfermedades sistémicas con marcada afectación
cutánea y músculo-esquelética y que en su patogenia
se encuentran alteraciones inmunes. En estas enfermedades se incluyen el
lupus eritematoso, la dermatomiositis y la esclerodermia.
La capacidad del sistema
inmune para distinguir lo propio de lo no propio es básico para generar las
reacciones de inmunidad que confieren protección frente a los patógenos así como
para mantener la tolerancia a los autoantigenos. Una alteración en el balance de
estas dos situaciones, es decir una ausencia de respuesta frente a patógenos
(inmunodeficiencia) o una respuesta excesiva o no controlada a antígenos
(alergia o autoinmunidad) pueden dar lugar a diversas manifestaciones
patológicas. El establecimiento de la autotolerancia representa un elemento
esencial para evitar la patogénesis de las enfermedades autoinmunes. La
supresión tímica de las células T autoreactivas es el mecanismo primario que da
lugar a la autotolerancia. Sin embargo esta selección de células T autoreactivas
no es completamente eficaz y en personas normales existen células T
autoreactivas. Sin embargo, las enfermedades autoinmunes no son muy frecuentes
(aunque pueden llegar a representar un 15% de las patologias en relación a la
definición de las mismas). La tolerancia inmunológica se mantiene por varios
mecanismos que incluyen la anergia, la ignorancia inmunológica y por mecanismos
activos regulados por las células T
reguladoras (Treg). A pesar de estos mecanismos se dan situaciones en que el
sistema inmune se dirige contra constituyentes propios del organismo, dando
lugar a las enfermedades autoinmunes.
A diferencia de
enfermedades autoinmunes órgano-específicas, como el pénfigo o la diabetes
mellitus insulinodependiente, las enfermedades del tejido conectivo, son un
grupo de enfermedades autoinmunes sistémicas que afectan a numerosos órganos y
tejidos. En estas enfermedades existe una falta de control de las células
T y B autoreactivas, dando lugar a la producción de autoanticuerpos dirigidos
contra autoantígenos que en su mayoría son de origen nuclear. El origen exacto
de esta pérdida de tolerancia no está bien entendido e incluye factores
intrínsecos y extrínsecos (predisposición genética, factores ambientales y
hormonales) con un posible papel de mecanismos como
mimetismo molecular entre un autoantigeno y un antígeno externo. La
activación no controlada de las células T y B también puede ser atribuida a una
función deficitaria de las células T reguladoras que controlan la activación
periférica de las células.
Lupus eritematoso
El lupus eritematoso (LE) es una enfermedad inflamatoria crónica
de base autoinmune, en la que se detectan autoanticuerpos y alteraciones
de la inmunidad celular dirigidos contra componentes tisulares normales -sin
especificidad para un órgano determinado-y caracterizada por el desarrollo de una gran variedad de manifestaciones
cutáneas y sistémicas. En las formas benignas, la enfermedad
puede estar limitada a las manifestaciones cutáneas, mientras que
en las formas severas -con o sin manifestaciones cutáneas-, predomina
la afectación sistémica. Para considerar que un enfermo de
lupus tiene la forma sistémica de la enfermedad debe cumplir al
menos 4 de los 11 criterios propuestos por la ARA y resumidos en la tabla III. El pronóstico de la forma sistémica de la enfermedad
ha mejorado mucho en los últimos años, con una supervivencia
a los 5 años por encima del 90%. Las complicaciones más importantes
en estos enfermos tienen un patrón bimodal, cuando se produce un
fallecimiento en los 2 primeros años del diagnóstico se debe
bien a una infección o a la actividad de la enfermedad, las muertes
producidas en fases tardías son frecuentemente debidas a complicaciones
cardiovasculares.
Video Dr. Ackerman
|
Tabla I
Lesiones cutáneas de lupus eritematoso
|
|
A. Lesiones especificas
|
B. Lesiones
inespecíficas
|
|
LE
cutáneo crónico
LE
cutáneo subagudo
LE
cutáneo agudo
-
Eritema malar
-
Eritema generalizado
|
Vasculopatia
-
Vasculitis
-
livedo reticulares
-
Fenómeno de Raynaud
-
Eritromelalgia
Lesiones ampollosas
Alopecia no cicatricial
Ulceras orales
Mucinosis cutánea |
|
Formas especiales de Lupus
|
Lesiones cutáneas del Lupus eritematoso
Las manifestaciones cutáneas en el curso de la enfermedad lúpica
son frecuentes y en muchas ocasiones son el motivo de consulta que orienta
hacia el diagnostico, algunas lesiones cutáneas se incluyen como
criterio diagnostico para clasificar una enfermedad lúpica como
sistémica.
Las lesiones cutáneas se clasifican en especificas o inespecíficas
en relación a si sus hallazgos clínico-patológicos
e inmunopatológicos son propios de la enfermedad lúpica.
Las lesiones inespecíficas son aquellas que además de observarse
en la enfermedad lúpica pueden observarse en otras entidades. La
clasificación de las lesiones cutáneas especificas y no especificas
esta resumida en la tabla I.
-
Lupus eritematoso cutáneo
crónico: La forma más
frecuente de lupus eritematoso cutáneo crónico es la forma
discoide o localizada, que consiste en el desarrollo de placas redondas
(discoides) u ovales, eritematosas, bien delimitadas, que aparecen en áreas
expuestas de la cara y frente, observándose en su superficie la
presencia de telangiectasias y escamas finas y adherentes. Es frecuente
observar en el centro de la lesión o en lesiones antiguas áreas
atróficas, áreas hipopigmentadas y lesiones de queratosis
folicular. Cuando existe afectación del cuero cabelludo se produce
una alopecia cicatricial. Existen formas en las que predomina la hiperqueratosis
denominándose LE hipertrófico o verrucoso. El lupus profundo
es otra forma de LE cutáneo crónico que se caracteriza
por la afectación del tejido graso dando lugar a una paniculitis
lúpica. El 50% de los pacientes con lupus profundo tienen lesiones
cutáneas superficiales asociadas, pero en el otro 50% solo existen
lesiones profundas, consistente en nódulos subcutáneos, indurados,
localizados frecuentemente en cara y porción proximal de extremidades.
Solo un 5% de pacientes con lupus eritematoso cutáneo crónico
cumple criterios de lupus sistémico.
-
Lupus eritematoso
cutáneo subagudo: El LE cutáneo
subagudo es otra forma de lupus cutáneo relativamente frecuente
que se observa en pacientes que refieren marcada fotosensibilidad. Es mas
frecuente en mujeres y las lesiones clínicas se desarrollan preferentemente
en la "V" del escote y áreas expuestas de extremidades. Las lesiones
clínicas son similares a las de LE crónico, pero en su evolución
no tienen tendencia a dejar cicatriz. Existen dos variedades importantes,
la forma anular y la forma psoriasiforme. La forma anular se caracteriza
por el desarrollo de placas anulares eritematosa con tendencia a la curación
central. La forma psoriasiforme se caracteriza por el desarrollo de placas
eritematosas con formación de escamas en su superficie. Un 50% de
los pacientes con lupus cutáneo subagudo van a cumplir criterios
de lupus sistémico, pero en general estos pacientes van a tener
una enfermedad poco grave, con marcada afectación articular y fotosensibilidad,
pero sin compromiso vital. Diversas medicaciones están siendo implicaddas en el
desarrollo de esta forma de lupus cutáneo, considerándose que desencadenan la
enfermedad en pacientes susceptibles de la misma posiblemente por mecanismos de
fotosensibilidad.
-
Lupus eritematoso
Agudo: La forma de
LE aguda es la que con mayor frecuencia se asocia al desarrollo de lupus
eritematoso sistémico. Se caracteriza por el desarrollo de rash
malar o en alas de mariposa, consistente en placas eritematosas, edematosas,
mal delimitadas afectando a ambas mejillas y dorso de nariz, con preservación
del pliegue nasolabial y región subnasal. Los pacientes con LE agudo
muestran marcada fotosensibilidad y ocasionalmente el debut de la enfermedad
se desarrolla con una quemadura solar intensa con desarrollo de ampollas.
Un 75% de los pacientes con esta forma de lupus van a cumplir criterios
de afectación sistémica. Este rash malar está presente en el
40-50% de pacientes en el momento del diagnóstico del lupus sistémico. Un tercio
de pacientes con lupus sistémico tienen una forma de luspus agudo generalizado,
con afectación predominante de las manos.
Histología
de las lesiones de lupus eritematoso cutáneo
|
Tabla II
Histología del Lupus eritematoso
|
-
Atrofia epidérmica e hiperqueratosis
-
Degeneración vacuolar de la capa basal epidérmica
con engrosamiento de la capa basal
-
Infiltrado inflamatorio linfocítico afectando a la
unión dermo-epidermica, anejos y perivascular
-
Incontinencia pigmenti
-
IFD: depósitos granulares de IgG, IgM y C3 en la membrana
basal
|
Las lesiones cutáneas
de lupus eritematoso especificas se acompañan de cambios histológicos
característicos a nivel de todas las estructuras cutáneas
incluyendo epidermis, dermis, anejos y tejido subcutáneo. En la
epidermis el cambio histológico más llamativo es la presencia
de una atrofia epidérmica con vacuolización de la capa basal,
que se acompaña de un engrosamiento de la membrana basal. En la
unión dermo-epidermica además del engrosamiento se puede
observar la existencia de una dermatitis de interfase con presencia de
infiltrado inflamatorio de predominio linfocítico que borra la unión
dermo-epidérmica, este infiltrado inflamatorio afecta además
de la unión dermo-epidermica a los anejos y en dermis suele acompañarse
de depósitos variables de mucina. En los anejos además de
la existencia del infiltrado inflamatorio es llamativa la existencia de
queratosis folicular. Inmunofluorescencia:
La IFD de las lesiones cutáneas específicas de lupus eritematoso
suele ser positiva mostrando la presencia de depósitos de IgG, IgM
y C3 granulares a nivel de la membrana basal. En pacientes con lupus eritematoso
sistémico se pueden observar depósitos de inmunoglobulinas
en piel aparentemente sana, lo que se conoce como el test de la banda lúpica,
que en la actualidad tiene escaso valor diagnóstico.
Valor de las lesiones cutáneas
en el diagnóstico de Lupus eritematoso sistémico
El diagnostico de lupus eritematoso
sistémico se basa en una serie de datos clínicos y analíticos
revisados por la ARA (Tabla III), debiéndose cumplir 4 de estos
11 criterios para clasificar al paciente como afecto de lupus sistémico.
Los primeros 4 criterios corresponden a manifestaciones dermatológicas
y son el desarrollo de rash malar, rash discoide, fotosensibilidad y ulceras
orales. El rash malar (lupus cutáneo agudo) es el primer criterio
diagnostico, es característico de la forma sistémica, y si
bien es cierto que su presencia hace sospechar la forma sistémica
su presencia aislada no es criterio suficiente para catalogar la enfermedad
como sistémica. El rash discoide (lupus cutáneo crónico)
se ha descrito en el apartado de las lesiones especificas de lupus eritematoso cutáneo
y generalmente se observa en pacientes con la enfermedad limitada a la
piel pero cerca de un 15% de los pacientes con LES tienen lesiones de discoide.
Las úlceras orales se observan en un 25-45% de pacientes con
LES y en un 25% de pacientes con lupus eritematoso crónico discoide,
histológicamente pueden mostrar cambios específicos de LE o ser inespecíficas,
debiéndose diferenciar de otras formas de
úlceras orales, como aftas, liquen plano, herpes simple etc. La
fotosensibilidad es un termino poco definido que comprende una mala
tolerancia al sol bien observada por el paciente o por el médico
o por una exacerbación de las manifestaciones cutáneas o
sistémicas de la enfermedad tras la exposición solar. En
2012 el grupo colaborativo internacional del lupus eritematoso sistémico hizo
una puesta al día el el sistema de clasificación del lupus sistémico,
incluyendo 11 criterios clínicos y 6 criterios inmunológicos, ampliando el
espectro de manifestaciones clínicas, especialmente cutáneas y neuropsiquiátricas
y con la inclusión de datos analíticos como los niveles bajos de complemento
como criterios de inclusión en el diagnóstico.
|
Tabla III
|
|
Criterios del ARA para el diagnostico de lupus eritematoso
sistémico |
Criterios para clasificación del LES degrupo clínico
internacional de lupus eritematoso sistémico
|
-
Rash malar
-
Rash discoide
-
fotosensibilidad
-
úlceras orales
-
Artritis no erosiva
-
Serositis (pleuritis o pericarditis)
-
Enfermedad renal (proteinuria >0,5 gr/día o presencia
de cilindros celulares o hemáticos)
-
Enfermedad neurológica (convulsiones o psicosis)
-
Enfermedad hematológica
-
Leucopenia (<4.000 en dos o más ocasiones)
-
Linfopenia (<1.500 en dos o más ocasiones)
-
trombocitopenia (<100.000)
-
Anemia hemolítica
-
Alteraciones serológicas
-
Anticuerpos anti-DNAds
-
Anticuerpos Anti-SM
-
Serología luética falsamente positiva,
-
anticuerpos antifosfolípidos (Ac Anticardiolipina
y/o anticoagulante lúpico) positivos
-
Anticuerpos antinucleares
|
Criterios
clínicos
-
Lupus
cutáneo agudo
-
Lupus
cutáneo crónico
-
Ulceras orales
-
Alopecia no cicatricial
-
Sinovitis en dos o mas articulaciones (incluyendo tumefacción y rigidez
matutina de 30 minutos)
-
Serositis
-
Renal: relación proteína/creatinina en orina o proteinuria de 24 horas
-
Neurológicas
-
Anemia hemolítica
-
Leucopenia
-
Trombocitopenia
Criterios
inmunológicos
-
Anticuerpos antinucleares
-
Anti
DNA de doble cadena
-
anti-Smith
-
Anticuerpos antifosfolípidos
-
Complemento bajo
-
Test
de coombs directo en ausencia de anemia hemolítica
|
|
Deben cumplirse 4 de estos 11 criterios para el
diagnóstico de lupus sistémico.
|
La clasificación necesita cuatro criterios
secuenciales, incluyendo un criterio clínico y un criterio inmunológico o una
biopsia de nefritis lúpica con anticuerpos antinucleares positivos o anti DNA de
doble cadena. Solo se valoran los criterios si no hay otra causa.
|
Lesiones cutáneas inespecíficas
Es importante reconocer las lesiones inespecíficas de cara a realizar el
diagnóstico de lupus eritematoso de forma precoz. Estas manifestaciones se
relacionan especialmente con el lupus eritematoso sistémico, con las posibles
complicaciones que ello comporta. La presentación de una
paciente joven con artritis inflamatoria y rash malar en alas de mariposa es
infrecuente. El diagnostico se estable con frecuencia con la presentación de
signos clínicos no específicos como el desarrollo de fatiga, malestar, úlceras
orales, artralgias, fotosensiblidad, rash cutáneo, fenómeno de Raynaud,
alopecia difusa, etc.
-
Vasculitis: Es la lesión cutánea inespecífica más
frecuente del lupus eritematoso sistémico, observándose entre
un 20 y 40% de los casos. La expresión clínica de las lesiones
de vasculitis es variable desde lesiones urticariformes, púrpura
palpable y úlceras cutáneas. Al igual que otras formas de
vasculitis la localización mas frecuente son las extremidades inferiores.
La mayoría de las vasculitis observadas en el contexto de un paciente
con LES son vasculitis mediadas por el depósito de inmunocomplejos. El estudio histológico de estas lesiones muestra una vasculitis
leucocitoclástica.
-
Livedo reticularis: Consiste en la existencia
máculas lineales azuladas, siguiendo un patrón vascular, dejando áreas pálidas
en su centro que les confieren un patrón en red, que no cambia significativamente con
las posturas ni con la temperatura, que se observa frecuentemente en extremidades
inferiores y glúteos, que puede acompañarse del desarrollo
de nódulos y/o úlceras, y que se ha relacionado con la presencia
de anticuerpos anticardiolipina y/o anticoagulante lúpico, pudiéndose
observar en otras entidades como el embolismo por colesterol.
-
Fenómeno de Raynaud: Se caracteriza por cambios vasculares
que afectan a dedos de manos y pies y consisten en tres fases, un
componente espástico inicial, doloroso, caracterizado por un blanqueamiento
asimétrico de los dedos, seguido de la aparición de una coloración
cianótica y un eritema tardío que refleja la reperfusión
de los dedos. Entre un 2 y un 10% de los pacientes con fenómeno
de Raynaud tienen LES, pero alrededor de un 30% de los pacientes con LES
presentan fenómeno de Raynaud. El fenómeno de Raynaud es
más frecuente en pacientes cuyo estudio serológico muestra
positividad para el anticuerpo anti-RNP(característico de la enfermedad
mixta del tejido conectivo).
-
Eritromelalgia: Consiste en el desarrollo de
brotes paroxísticos de eritema, aumento de la temperatura, sensación
de quemazón y dolor intenso afectando a palmas y/o plantas. Suele
ser secundario a procesos mieloproliferativos, como trombocitemia
o policitemia vera, pero en ocasiones se observa en pacientes con LES.
-
Alopecia: Los pacientes con lupus pueden
además de desarrollar alopecia cicatricial por afectación
del folículo por lesiones de lupus discoide, desarrollar alopecia
difusa -en general efluvio telógeno- asociada al proceso inflamatorio
sistémico.
Formas especiales de lupus
eritematoso
-
Lupus
tumidus: Este
es un subtipo de lupus cutáneo relativamente frecuente que se caracteriza por el
desarrollo de lesiones cutáneas en forma de placas eritemato-edematosas, de
curso agudo, que aparecen en áreas expuestas (especialmente cara y escote),
asociadas a marcada fotosensibilidad y que a diferencia de otras formas de lupus
cutáneo no se acompaña de afectación epidermica en forma de atrofia y/o
queratosis folicular, curando las lesiones sin dejar cicatriz. El lupus tumidus
suele afectar más frecuentemente a varones de mediana edad, tiene un curso
benigno despareciendo las lesiones espontáneamente o con tratamiento,
presentando frecuentes recidivas en las épocas estivales. Tiene una baja
incidencia de afectación sistémica.
-
Lupus Neonatal: Es un síndrome que aparece
en recién nacidos consecuencia del paso trasplacentario de anticuerpos
maternos al niño, especialmente Anti Ro y La. Se caracteriza por
presentar alteraciones cutáneas, hematológicas y viscerales.
Las lesiones cutáneas son transitorias, se exacerban con la exposición
solar y son similares a las observadas en en lupus eritematoso subagudo
consistiendo en máculas eritematosas, anulares y poli cíclicas
que con frecuencia afectan a la región periorbitaria. La lesión
sistémica más importante es el desarrollo de bloqueo aurículo-ventricular
que se observa en un 5% de hijos de madres anti-Ro positivas y que suele
ser irreversible requiriendo la implantación de un marcapasos en
el 60-70% de los casos y teniendo una mortalidad que se ha descrito de
hasta el 30% de casos.
-
 Síndrome de anticuerpos
antifosfolípidos: Es un síndrome caracterizado
por el desarrollo de trombosis venosas profundas, trombosis arteriales,
tombocitopenia y abortos de repetición asociado a la presencia de anticuerpos
anticardiolipina, anticoagulante lúpico o anticuerpos anti
β2-glicoproteina
I. El síndrome
de anticuerpos antifosfolípidos puede ser primario (>50% casos) o secundario
según esté asociado a otras enfermedades del tejido conectivo,
siendo la más frecuente el lupus eritematoso sistémico (entre el 20 y el 35% de
pacientes con lupus sistémico desarrollaran un síndrome de anticuerpos
antifosfolipídico secundario). Existe una variante
-Síndrome
antifosfolípidico catastrófico-
que da lugar a una microangiopatía trombótica diseminada y fallo multiorgánico. Las lesiones
cutáneas observadas en pacientes con síndrome de anticuerpos
antifosfolípidos incluyen el desarrollo de úlceras cutáneas, livedo reticularis, vasculitis livedoide y tromboflebitis superficial.
El estudio histológico de las lesiones cutáneas muestra la
presencia de una trombosis no inflamatoria de los vasos dérmicos.
Los anticuerpos antifosfolípidos pueden detectarse analíticamente
de 3 maneras: 1) anticuerpos
anticardiolipina, 2)Anticoagulante lúpico en plasma en dos o más ocasiones y 3)
anticuerpos anti
β2-glicoproteina
I, presentes en más de 2 ocasiones.; la medición
de los anticuerpos anticardiolipina es el test más sensible para
detectar su presencia, pero pueden ser positivos en otras enfermedades
autoinmunes, infecciones e incluso en la población normal.
Síndrome de anticuerpos
antifosfolípidos: Es un síndrome caracterizado
por el desarrollo de trombosis venosas profundas, trombosis arteriales,
tombocitopenia y abortos de repetición asociado a la presencia de anticuerpos
anticardiolipina, anticoagulante lúpico o anticuerpos anti
β2-glicoproteina
I. El síndrome
de anticuerpos antifosfolípidos puede ser primario (>50% casos) o secundario
según esté asociado a otras enfermedades del tejido conectivo,
siendo la más frecuente el lupus eritematoso sistémico (entre el 20 y el 35% de
pacientes con lupus sistémico desarrollaran un síndrome de anticuerpos
antifosfolipídico secundario). Existe una variante
-Síndrome
antifosfolípidico catastrófico-
que da lugar a una microangiopatía trombótica diseminada y fallo multiorgánico. Las lesiones
cutáneas observadas en pacientes con síndrome de anticuerpos
antifosfolípidos incluyen el desarrollo de úlceras cutáneas, livedo reticularis, vasculitis livedoide y tromboflebitis superficial.
El estudio histológico de las lesiones cutáneas muestra la
presencia de una trombosis no inflamatoria de los vasos dérmicos.
Los anticuerpos antifosfolípidos pueden detectarse analíticamente
de 3 maneras: 1) anticuerpos
anticardiolipina, 2)Anticoagulante lúpico en plasma en dos o más ocasiones y 3)
anticuerpos anti
β2-glicoproteina
I, presentes en más de 2 ocasiones.; la medición
de los anticuerpos anticardiolipina es el test más sensible para
detectar su presencia, pero pueden ser positivos en otras enfermedades
autoinmunes, infecciones e incluso en la población normal.
-
Lesiones ampollas y
lupus ampolloso: Los pacientes con lupus pueden
desarrollar ampollas cutáneas o mucosa de diversa etiología:
El desarrollo de ampollas puede observarse en el curso de un lupus cutáneo
crónico o subagudo en el que exista una marcada afectación
vacuolar de la capa basal que de lugar a la separación dermo-epidermica,
pudiéndose observar también tras una exposición solar
intensa especialmente en pacientes con anticuerpos Anti-Ro. También
puede observarse en pacientes con LES el desarrollo de una enfermedad ampollosa
autoinmune asociada a la presencia de anticuerpos circulantes contra antígenos
de 290 y 145 kD, similares a los observados en la epidermolisis ampollosa
adquirida, que son constituyentes del colágeno VII, localizados
en la sublámina densa y que asocia el desarrollo de ampollas subepidérmicas.
Raramente los pacientes con lupus eritematoso pueden asociar otra enfermedad
ampollosa autoinmune o de otra etiología.
Estudios serológicos en el lupus eritematoso
Los pacientes con lupus eritematoso presentan en suero diversos autoanticuerpos
dirigidos contra diversos antígenos nucleares incluyendo DNA,
RNA y otras proteínas, algunos de estos anticuerpos pueden observarse
en varias enfermedades del colágeno mientras que otros son
específicos de la forma sistémica de la enfermedad o se asocian
con alguna de las variantes. Los anticuerpos antinucleares presentes en
el lupus están resumidos en la tabla IV.
|
Tabla IV
Anticuerpos antinucleares
|
|
ANA |
homogéneo |
Anti-DNA, antihistonas |
|
Periférico |
Anti-DNA nativo |
|
Moteado |
Anti ENA (RO, LA, SM, U1RNP) |
|
nucleolar |
Ac anti proteínas y RNA nucleolar |
|
Anti
DNA |
Anti-DNAss |
Se observan en el 70% de pacientes
con LES y también en otras enfermedades del tejido conectivo |
|
Anti-DNAds |
presentes en el 28% de pacientes
con LES, son muy específicos y su nivel se relaciona con la actividad
de la enfermedad |
|
Anti-DNAss + Anti DNAds |
presentes en el 70-80% de pacientes
con LES |
|
Anti
ENA |
Anti-SM |
se observan en el 25% de pacientes
con LES, son patognomónicos |
|
Anti RNP |
aparecen en 25-30% de pacientes
con LES con manifestaciones de Raynaud, también se observan en la
enfermedad mixta de tejido conectivo |
|
Ac Anti Ro |
50-100% casos de LECS, Lupus neonatal
y S. Sjogren |
|
Ac Anti La |
Casi siempre van asociados a anti-Ro, son
característicos del síndrome de Sjogren observándose
también en el lupus neonatal, enfermedad mixta y en el 10% de LES |
Anticuerpos antinucleares:
Los anticuerpos antinucleares son resultado de una reacción de autoinmunidad
que aparece en sujetos genéticamente predispuestos, son inmunoglobulinas
(predominantemente IgG) dirigidas contra moléculas nucleares bien
de DNA, RNA o proteínas nucleares, nucleolares y/o citoplásmicas.
Los anticuerpos antinucleares se detectan mediante una técnica de
inmunofluorescencia indirecta utilizando como sustrato cortes congelados
de hígado de rata, esta técnica puede presentar
4 patrones: 1)Patrón homogéneo o difuso: se observa
en pacientes con anticuerpos antihistonas y anti DNA, no es especifico
de LES y se ha descrito en otras enfermedades autoinmunes, 2) Patrón
periférico: es muy específico de LES y detecta anticuerpos
anti-dsDNA, 3)patrón moteado: traduce la existencia de anticuerpos
frente a diferentes proteínas no histonas extraíbles del
núcleo (ENA: Ro, La y U1RNP) 4) patrón nucleolar:
solo se tiñen los nucleolos de las células e indica la presencia de anticuerpos contra el ARN nucleolar, siendo su incidencia muy baja en
el LE, observándose en pacientes con esclerodermia. Si bien la positividad
de los ANA es criterio diagnóstico de LES (están presentes
en el 98-100% de pacientes), se considera un marcador de enfermedad autoinmune
no específica de LES ya que también pueden encontrarse en
otras enfermedades del tejido conectivo tales como Enfermedad mixta
del tejido conectivo (95-100% casos), Esclerodermia sistémica (70-96%),
Síndrome de Sjogren (70-90%), Polimiositis (70-80%), artritis reumatoide
(30-60%) e incluso un 5-10% de la población normal presenta ANA,
si bien a títulos bajos y generalmente de clase IgM.
Anticuerpos Anti-DNA:
Son anticuerpos dirigidos contra la cadena de DNA pueden ser de 3 tipos
dirigidos contra DNA de cadena única (DNAss), también denominado
DNA desnaturalizado; dirigidos contra DNA de doble cadena (DNA-ds), también
llamado DNA nativo, y dirigidos contra determinantes del DNA
presentes tanto en el DNA-ss o DNA-ds. Los anticuerpos anti DNA están
presentes en el 40-70% de pacientes con LES, siendo infrecuente en otras
enfermedades autoinmunes. Si los niveles son elevados suele ser específico
de lupus sistémicos y a niveles muy elevados suele correlacionarse
con la actividad clínica y sugerir la existencia de una enfermedad
renal subyacente.
Anticuerpos anti-ENA:
Los anticuerpos anti-ENA son un grupo de anticuerpos muy frecuentes en
las enfermedades autoinmunes, dirigidos contra antígenos nucleares
y citoplásmicos solubles, incluyen varias estructuras antigénicas
diferentes -SM, RNP, Ro, La- y aparecen en el 10-40% de los pacientes con
LES, detectándose también en otras enfermedades autoinmunes.
-
Anticuerpos Anti-SM
: se observan en el 20-25% de pacientes con LES y su presencia es muy específica
de LES, su presencia se asocia con el desarrollo de nefritis lupica, afectación
del sistema nervioso central, pulmonar y pericardio.
-
Anticuerpos Anti-RNP
: dirigidos contra una proteina nuclear. Estos anticuerpos (especialmente
el anti-U
1 RNP) son muy característicos de la enfermedad
mixta del tejido conectivo. Un 20-30% de pacientes con LES presentan estos
anticuerpos.
-
Anticuerpos Anti-Ro:
Los anticuerpos anti-Ro están dirigidos contra ARN citoplásmico
y su presencia se relaciona con manifestaciones cutáneas características
de lupus cutáneo subagudo con marcada fotosensibilidad y en general
con poca afectación sistémica. También es característica
su presencia en el lupus neonatal, y síndrome de solapamiento entre
lupus cutáneo subagudo y síndrome de Sjogren.
-
Anticuerpos Anti La:
casi siempre se encuentran asociados a los anticuerpos Anti-Ro y se observan
en el síndrome de Sjogren, y en las madres de niños con lupus
neonatal, su presencia en el LES al igual que los anti-Ro confiere un buen
pronóstico.

|
Tabla V
Tratamiento del lupus eritematoso cutáneo
|
-
Estándar
-
Educación del paciente
-
Fotoprotección
-
Corticoides tópicos
-
Antimaláricos
-
Sulfato de cloroquina 250 mg/12-24 h (Es necesario realizar
examen oftalmológico antes del inicio del tratamiento y cada 6 meses
así como niveles de la enzima Glucosa 6-fosfato deshidrogenasa)
-
Hidroxicloroquina
-
mepacrina
-
Tratamiento en casos resitentes
-
Corticoides sistémicos
-
Inmunosupresores
-
Talidomida
-
Sales de oro
|
Tratamiento del lupus eritematoso cutáneo :
El tratamiento de las lesiones cutáneas de lupus (tabla V) incluye medidas
locales, mediante la utilización de fotoprotección y de corticoides
tópicos. El tratamiento sistémico se realiza mediante la
administración de antimaláricos (sulfato de cloroquina o
hidroxicloroquina), siendo preciso realizar controles oftalmológicos
periódicos para descartar el desarrollo de retinopatía. La eficacia de los antimaláricos se ve disminuida por el tabaquismo.
Dermatomiositis
La dermatomiositis es una enfermedad inflamatoria, de probable base autoinmune,
que se caracteriza por la afectación muscular con afectación
predominante de las cinturas musculares pelvianas y escapulares, por el
desarrollo de lesiones cutáneas características y por la
posible afectación de órganos internos. El término
de polimiositis se utiliza en aquellos pacientes en los que no existe afectación
cutánea.
La polimiositis y dermatomiositis
son clasificadas como una misma enfermedad con un espectro clínico
diferente, clasificándose en 6 tipos según las manifestaciones
y características clínicas (tabla VI).
La dermatomiositis es relativamente infrecuente, con una incidencia anual
de 5-10 casos por millón y año y una prevalencia de 5-8 casos
por cada 100.000 habitantes, es más frecuente en mujeres y se puede
manifestar en cualquier edad, existiendo dos picos de mayor incidencia
uno en la infancia (<10 años) y otro en la edad adulta (45-60
años). El diagnóstico de la dermatomiositis debe establecerse
según unos criterios diagnósticos que son útiles
para la realización de estudios y que están resumidos en
la tabla VII.
|
Tabla VI
Clasificación de la dermatomiositis/polimiositis
|
|
I |
Polimiositis idiopática
primaria |
|
II |
Dermatomiositis idiopática
primaria |
|
III |
Polimiositis o dermatomiositis asociada
a neoplasia |
|
IV |
Dermatomiositis o polimiositisjuvenil |
|
V |
Polimiositis o dermatomiositis asociada a
otra enfermedad de tejido conectivo (Síndrome de solapamiento) |
|
VI |
Dermatomiositis amiopática |
|
|
Tabla VIII Manifestaciones
clínicas de la dermatomiositis/polimiositis
|
|
Cutáneas |
Poiquiloderma
Pápulas
de Gottron
Rash heliotropo
Engrosamiento
de la cutícula
Manos de mecánico
Fenómeno
de Raynaud |
|
Músculo-esqueleticas |
Artralgias y
artritis
Debilidad muscular
de cintura escapular y pelviana
|
|
Gastrointestinales |
Disfagia con regurgitación nasal
Perforación
del tracto gastrointestinal secundaria a vasculitis (dermatomiositis juvenil) |
|
Pulmonares |
Reducción
de la capacidad ventilatoria secundaria a la afectación de músculo
diafragmático y músculos accesorios
Neumonía
por aspiración
Infecciones
secundarias a la terapia
Fibrosis intersticial
|
|
Cardíacas |
Miocarditis
sintomática infrecuente asociada con arritmias y insuficiencia cardíaca
congestiva
Cor pulmonale
secundario a enfermedad pulmonar intersticial
|
|
|
Tabla VII
Criterios diagnósticos de DMS de Bohan y Peter
|
-
Debilidad simétrica proximal, progresando
durante semanas o meses
-
Biopsia muscular que evidencia una miopatia
inflamatoria
-
Aumento de los niveles séricos de enzimas
musculares
-
Hallazgos electromiográficos de miopatia
-
Erupción cutánea típica de dermatomiositis
|
|
PM/DM definitiva: 4 criterios
PM/DM probable: 3 criterios
PM/DM posible: 2 criterios
Criterio 5 necesario para considerar DM |
|
Manifestaciones clínicas:
La dermatomiositis/polimiositis tiene una expresión clínica
variada que está resumida en la tabla VIII.
Manifestaciones
cutáneas
|
Manifestaciones cutáneas de la dermatomiositis |
|
Patognomónicas |
Pápulas de Gottron |
Pápulas y placas violáceas,
con áreas descamativas en dorso de articulaciones interfalángicas y
metacarpofalángicas |
|
|
Signo de Gottron |
Maculas eritematosas en las
áreas de extensión de codos, rodillas, tobillos y nudillos |
|
|
Rash heliotropo |
Eritema periorbitario con
edema, afectando con frecuencia a párpados superiores |
|
Características |
Cambios en la cutícula |
Eritema periungueal con
telangiectasias, cutículas distróficas e infartos hemorrágicos en el
lecho ungueal |
|
|
Signo del chal |
Maculas y placas
eritematosas o violáceas en la porción posterior de los hombros,
cuello, espada y cara lateral de brazos |
|
|
Signo V |
Maculas eritematosas
confluentes en la porción inferior del cuello |
|
|
Signo de Holster |
Poiquilodermia de caderas y
cara lateral de muslos por debajo del trocánter mayor |
|
|
Afectación del cuero
cabelludo |
Placas descamativas
ocasionalmente pruríticas y atróficas |
-
Pápulas y signo de Gottron: Se observan en más del 80% de pacientes con dermatomiositis
y se consideran patognomónicas de la enfermedad. Consisten en pápulas y
placas violáceas localizadas en la cara dorsal de las articulaciones
interfalángicas y metacarpofalángicas. Se considera el signo de
Gottron la presencia de un eritema macular violáceo en la misma localización
y que también puede verse localizado alrededor de otras articulaciones
incluyendo codos, rodillas, tobillos.
-
Rash heliotropo
:Se observa hasta en el 60% de los pacientes consiste en el desarrollo de
áreas de poiquilodermia afectando a párpados y región periorbitaria, con
marcada fotosensibilidad, ocasionalmente se asocia con la presencia de
marcado edema, especialmente cuando se observa en una dermatomiositis
asociada a neoplasia.
-
Poiquilodermia:Constituye un hallazgo clínico cutáneo muy característico
de la dermatomiositis. Consiste en el desarrollo de placas cutáneas
con áreas hiper e hipopigmentadas, atróficas y con telangiectasias
prominentes, con marcada fotodistribución, afectando a la región
de la "V" del escote (signo del chal) y base del cuello.
-
Afectación de la cutícula : La cutícula periungueal se puede observar engrosada con
presencia de telangiectasias que a simple vista o mediante capilaroscopia
mediante oftalmoscopio puede demostrar la presencia de pequeños
vasos capilares de trayecto tortuoso. También pueden observarse
áreas de hemorragia en astilla e infarto.
-
Manos
de Mecánico : consiste en el desarrollo de áreas
de hiperqueratosis y fisuración de las caras laterales y palmares
de los dedos de las manos, lo cual produce un aspecto de manos sucias similares
a las observadas en los mecánicos.
-
Calcinosis
cutis: constituye una complicación tardía que se
observa en el 15% de las dermatomiositis de los adultos y en el 60% de
dermatomiositis juvenil. Consiste en el deposito de calcio en el
tejido subcutáneo y fascia y puede dar lugar al desarrollo de ulceras
cutáneas.
-
Otros hallazgos: Los pacientes con
dermatomiositis pueden desarrollar varios tipos de lesiones cutáneas
características de la enfermedad pero no patognomónicas que incluyen la
presencia de pápulas hiperqueratósicas foliculares de distribución lineal
afectando al dorso de manos, vasculitis y fenómeno de Raynaud.
Histología de las
lesiones cutáneas: El estudio histológico de las lesiones
cutáneas de la dematomiositis muestra cambios similares a los observados
en en lupus eritematoso, en las pápulas de Gottron existe una acantosis
epidérmica y engrosamiento marcado de la membrana basal que adquiere
un aspecto tortuoso con marcada degeneración vacuolar de la membrana
basal que en ocasiones puede acompañarse de intensos depósitos
de fibrina en la unión dermo-epidérmica. En dermis existe
un infiltrado inflamatorio intersticial de predominio linfocítico
acompañado de depósitos de mucina y ocasionalmente pueden
observarse focos de calcificación.
Afectación sistémica
-
Afectación muscular:
El desarrollo de alteraciones clínicas y analíticas
de afectación muscular es un hallazgo frecuente en pacientes
con dermatomiositis. Las lesiones cutáneas preceden a la afectación
muscular en la mayoría de pacientes con dermatomiositis. La afectación
muscular afecta principalmente a los músculos de la cintura escapular
y pelviana y generalmente tiene una distribución simétrica.
Los pacientes suelen referir debilidad y fatiga así como dificultad
o incapacidad para realizar esfuerzos a los que normalmente estaban habituados
tales como subir escaleras, levantar los brazos para peinarse o afeitarse,
levantarse de una silla, etc. La afectación muscular puede ser progresiva
con desarrollo de dificultad en deglución y afectación
de los músculos respiratorios. La afectación clínica
muscular se acompaña de alteraciones analíticas, electromiográficas
y patológicas en las biopsias musculares. Analíticamente
se pueden observar elevaciones de enzimas como la CPK, LDH, aldolsa
y transaminasas, pero no necesariamente han de estar todas ellas elevadas.
La medición de niveles de CPK es probablemente la más útil
para determinar la afectación muscular. En pacientes con enzimas
normales, la medición de excreción urinaria de creatinina
puede reflejar la actividad inflamatoria muscular.
Los estudios electromiograficos
son característicos mostrando una innervación nerviosa intacta,
lo que es útil para descartar procesos neuropáticos.
Las alteraciones electromiográficas características consisten
en la presencia de potenciales miopáticos caracterizados por unidades
o potenciales de acción polifásicos de baja amplitud o voltaje
y de corta duración bajo activación voluntaria, así
como un incremento de la actividad espontánea con fibrilaciones,
descargas complejas repetitivas y ondas agudas positivas.
La biopsia muscular debe realizarse
de músculos clínicamente afectos y va a mostrar un infiltrado
inflamatorio endomisial compuesto predominantemente por linfocitos
que se distribuyen preferentemente en las áreas perivasculares de
los septos interfasciculares. Pueden observarse grados variables de necrosis
de fibras en la periferia de los haces musculares. Otras técnicas exploratorias
útiles en la valoración de la afectación muscular
incluye la resonancia magnética nuclear y resonancia magnética
espectroscópica con fósforo 31, que pueden ser útiles
en detectar la afectación muscular y que en el futuro puede que
sustituyan a la electromiografía y a la biopsia muscular.
-
Afectación pulmonar:
un 15-30% de pacientes tienen afectación pulmonar en forma de fibrosis
intersticial difusa, que se observa especialmente en pacientes con anticuerpos
antisintetasa. Los pacientes con dermatomiositis también pueden
desarrollar neumonía por aspiración o insuficiencia respiratoria
si existe afectación de la musculatura intercostal o diafragmática.
|
Tabla IX.
Anticuerpos en la Dermatomiositis
|
|
Ac Inespecíficos |
Ac antinucleares |
+ a títulos bajos, en síndromes
de solapamiento se observan a títulos >1:160 |
|
Ac específicos |
Ac anti-sintetasa (Anti Jo, OJ,
PL, KU, KS) |
Su presencia se asocia al síndrome
antisintetasa |
|
Ac Anti Mi-2 |
se asocia con el desarrollo de
dermatomiositis |
|
Ac Anti 155/140 |
Asociado a una alta relación con neoplasia
También asociado a dermatomiositis juvenil |
|
Ac Anti PM/SCL |
Asociación PM/esclerodermia,
esclerodermia solo |
|
Ac asociados a DM |
anti-RNP |
Enfermedad mixta del tejido conectivo |
|
Anti-Ku |
Síndrome de solapamiento
PM-SCL, SLE, esclerodermia |
Estudios serológicos en pacientes con Dermatomiositis
Los pacientes con dermatomiositis muestran positividad para una gran variedad
de anticuerpos (Tabla IX). Los anticuerpos encontrados en estos pacientes pueden
clasificarse en los asociados a miositis que también se encuentran en otras
enfermedades autoinmunes y los específicos de miositis que se encuentran
especialmente en este grupo de enfermos. Un 90% de pacientes muestra positividad para
los ANA generalmente a títulos bajos. Un 20% muestran anticuerpos
anti -Mi-2, dirigido contra un antígeno nuclear de 240-kD, que muestra
una alta especificidad par la dermatomiositis (el 95% de pacientes que
lo presentan tienen dermatomiositis). Los pacientes con polimiositis y
menos frecuentemente los afectos de dermatomiositis presentan anticuerpos
contra diferentes tipos de aminoacil-tRNA sintetasas, que constituyen un
grupo de enzimas que unen la tRNA a el aminoácido correspondiente
(catalizan la formación de aminoacil-tRNA), estos anticuerpos tienen
una alta especificidad para el complejo polimiositis/dermatomiositis, el
más frecuente es el anti -Jo-1 que se observa en un 20% de pacientes
de polimiositis, en un 60-70% de pacientes que tienen además fibrosis
pulmonar intersticial y en un pequeño porcentaje (5%) de pacientes
con dermatomiositis. La presencia de estos anticuerpos define un síndrome
"síndrome antisintetasa", caracterizado por la presencia de enfermedad
pulmonar intersticial, disnea, manos de mecánico, artritis, fiebre,
fenómeno de Raynaud, síndrome del túnel carpiano,
polimiositis y dermatomiositis. Los anticuerpos anti-sintetasa casi nunca
se observan en pacientes con dermatomiositis asociada a neoplasia ni en
dermatomiositis juvenil. Dentro de los anticuerpos musculo-específicos, se ha
demostrado que la presencia de anticuerpos contra las proteínas de 155/140 KD
se han relacionado con una alta incidencia de cáncer y también se han demostrado
en los pacientes con dermatomiositis juvenil. Existe un grupo de anticuerpos que se han detectado
en enfermos con síndrome de solapamiento entre miositis-esclerodermia
que incluyen los anticuerpos anti PM-SCL, anti -Ku y anti RNP.
Subgrupos de Dermatomiositis
-
Dermatomiositis
juvenil
La dermatomiositis juvenil es una forma de miositis inflamatoria acompañada
de lesiones cutáneas que se observa en la población infantil
y juvenil (2-15 años). Las lesiones cutáneas así como
la afectación muscular son de intensidad variable y semejantes a
las de la DMS/PM de la población adulta, pero en general tiene un
mejor pronóstico. Las lesiones cutáneas más frecuentes
son el desarrollo del rash heliotropo periorbitario y en extremidades,
eritema periungueal, lesiones psoriasiformes en cuero cabelludo. Este grupo
de pacientes desarrolla calcificaciones y vasculitis con mayor frecuencia
que la población adulta. Las calcificaciones se observan en un 30-70%
de los casos y se observan en codos, rodillas nalgas y zonas traumatizadas
pudiendo afectar al tejido subcutáneo, muscular, fascia y exosquelético.
Las vasculitis pueden afectar a piel, músculo y tubo digestivo donde
se expresan clínicamente en forma de abdomen agudo.
-
Dermatomiositis
amiopátíca: describe un grupo de pacientes con lesiones
cutáneas características de dermatomiositis que tras un período
de observación no presentan manifestaciones musculares, clínicas, histológicas o con técnicas de imagen (MRI).
Representa alrededor del 10% de pacientes con dermatomiositis, afecta predominantemente
a adultos jóvenes. La lesión cutánea más característica
es el desarrollo de rash heliotropo periorbitario, los pacientes también
suelen tener lesiones cutáneas acrales con pápulas y signo
de Gottron así como telangiectasias periungueales. Es frecuente
el hallazgo de fotosensibilidad marcada, artralgias y fenómeno de
Raynaud. En este grupo de pacientes suele referir manifestaciones sistémicas
en forma de mal estado general y cansancio, pero los estudios clínicos
descartan la afectación muscular. Alrededor del 50% de los pacientes
tienen anticuerpos contra CADM-140 (también denominado anti -MDA-5) lo que ayuda
a establecer el diagnóstico.
-
Dermatomiositis asociada
a enfermedades del tejido conectivo: Alrededor de un 20% de pacientes
con DM/PM presentan signos de otras enfermedades del tejido conectivo
tales como LES, esclerodermia, S. Sjogren, AR, o enfermedad mixta del tejido
conectivo, presentando manifestaciones comunes a ambas entidades. Estos
pacientes suelen presentar una miositis leve. Serologicamente tienen una
mayor tendencia a presentar anticuerpos asociados a la dermatomiositis,
pero no especificos, con unos ANA de más de 1:640, anti DNAds, anti
ENA( especialmente anti RNP). Los pacientes que presentan el anticuerpo
anti PM/SCL engloban un grupo de pacientes que presentan signos clínicos
de esclerodermia y dermatomiositis que se han descrito bajo el término
de escleromiositis. Son niños que refieren miositis y mialgias,
con presencia de dedos escleróticos y fenómenos de Raynaud.
-
Dermatomiositis y neoplasia
maligna: Diversos estudios retrospectivos han descrito la asociación
entre dermatomiositis y cáncer con una frecuencia que oscila entre
un 10 y un 50%. Las neoplasias se observan en asociación con la dermatomiositis del adulto pero no el la forma juvenil ni en la polimiositis.
Ciertos tipos de neoplasia como el carcinoma de ovario en las mujeres y gástrico o linfomas en los varones aparecen más frecuentemente
en pacientes con dermatomiositis que en la población normal. En
pacientes de más de 45 años con DMS deben realizarse estudios
clínicos, analíticos y radiológicos para descartar
la presencia de una neoplasia.
Tratamiento de la dermatomiositis:
La administración sistémica de corticoides es el tratamiento
inicial de la dermatomiositis, si la afectación muscular no es muy
importante, la administración de dosis bajas (<0,5 mg/kg/d) se
relaciona con menos efectos secundarios. En pacientes en que no se controla
la enfermedad deben asociarse otros inmunosupresores como el metotrexate
y azatioprina, ciclosporina o inmunoglobulinas endovenosas.
Esclerodermia:
La esclerodermia es una enfermedad del tejido conectivo que afecta de forma
primaria a la piel y al tejido subcutáneo y que puede afectar a
órganos sistémicos como los pulmones, tracto digestivo y
corazón y cuyo hallazgo patológico más característico
es el depósito en exceso de colágeno dando lugar a fibrosis
en los tejidos afectos.
La esclerodermia se clasifica
en formas localizadas con afectación únicamente cutánea
y formas sistémicas en las que existe afectación visceral (Tabla X). El término de esclerodermia se aplica tanto a la enfermedad
localizada como a la sistémica, si bien también suele utilizarse
el término de morfea para las lesiones localizadas y el término
de esclerodermia o esclerosis sistémica para las formas sistémicas.
Las dos enfermedades difieren tanto en la forma de evolución como
en el pronostico. Los cambios cutáneos que presentan son similares
en forma de esclerosis cutánea y de atrofia. La relación
entre la esclerodermia localizada, o morfea, y la esclerodermia sistémica
es semejante a la existente entre el lupus eritematoso cutáneo y
el sistémico.
|
Tabla X
Formas clínicas
de esclerodermia |
|
Localizada |
Morfea |
Localizada |
Placas únicas o múltiples localizadas
preferentemente en el tronco |
|
Generalizada |
Cuatro o más placas de >3 cm, afectando >2 áreas
corporales.Lesiones generalizadas, sin afectación
visceral ni fenómeno de Raynaud |
|
Lineal |
Tronco y extremidades |
Afectación preferente en la infancia.
Los cambios cutáneos siguen una distribución dermatomal y
pueden provocar cambios en el crecimiento |
|
cabeza y cuello |
En golpe de sable |
Induración lineal de la dermis de cara y cuero
cabelludo, con posible afectación de músculo, hueso y sistema nervioso central |
|
Parry Romberg |
Pérdida de dermis, tejido subcutáneo, músculo y
hueso con afectación facial unilateral |
|
Morfea panesclerótica |
Afectación circunferencial de miembros con
afectación de epidermis, dermis, tejido subcutáneo, músculo y hueso. |
|
Mixta |
combinación de 2 o más tipos de morfea localizada |
|
Sistémica |
Esclerodermia sistémica limitada |
esclerosis cutánea acral (distal de
muñecas o tobillos). Fenómeno de Raynaud de mucho tiempo
de evolución |
|
Esclerodermia sistémica difusa |
Afectación acral y troncal. Fenómeno
de Raynaud y cambios cutáneos de poco tiempo de evolución |
|
Síndromes Overlap |
Hallazgos clínicos de esclerodermia
sistémica y al menos de una de otra enfermedad autoinmune (lupus,
dermatomiositis y/o artritis reumatoide) |
Esclerodermia
localizada o morfea:Es una enfermedad benigna de curso crónico,
caracterizada por la presencia de áreas de piel indurada. La morfea es de causa
desconocida. Clínicamente existen 2 formas de morfea, la llamada morfea en
placas (localizada y generalizada) y la morfea lineal.
La morfea en placas se caracteriza por el desarrollo de placas eritematosas, que
en el centro de la lesión adoptan una coloración blanquecina mientras que en los
bordes están rodeadas de un anillo eritemato-violáceo, conocido como anillo
lileáceo. A medida que la lesión progresa, la zona blanquecina central se hace
más evidente, con áreas de endurecimiento cutáneo, desaparición de los anejos y
cambios pigmentarios. Las placas suelen ser redondas u ovaladas de un tamaño
entre 2 y 15 cm.
La morfea lineal es una forma de morfea en la cual las lesiones se distribuyen
longitudinalmente afectando a una extremidad y suelen asociarse a limitaciones
en el movimiento. Existe una forma especial de esclerodermia lineal denominada
"esclerodermia en golpe de sable", en la cual las lesiones afectan inicialmente
a la región frontoparietal con la aparición de áreas de endurecimiento y
contracción cutánea, que pueden asociarse a alopecia cicatricial y atrofia ósea
subyacente.
Otra forma rara de morfea es la morfea panesclerótica incapacitante de los niños, en la cual no
existe afectación sistémica. Se caracteriza por una marcada
esclerosis superficial y profunda que afecta principalmente a tronco y
extremidades, con desarrollo de contracturas en flexión, osteoporosis,
artralgia y deformidades en garra de las manos.
El estudio histológico
de las lesiones de morfea se caracteriza por una atrofia epidérmica
y por un engrosamiento dérmico con presencia de una colágena
engrosada y densa, con aumento del número de fibroblastos y pérdida
de los anejos cutáneos.
Esclerodermia sistémica
La esclerodermia sistémica
es una enfermedad del tejido conectivo en la que existe afectación
cutánea y de órganos internos incluyendo el tracto digestivo,
pulmones, riñones y corazón. Se desconoce la etiología
exacta de la esclerodermia, dado que se asocia a la presencia de autoanticuerpos
y alteraciones en la inmunidad humoral y celular, se clasifica dentro de
las enfermedades autoinmunes, pero también existe la posibilidad
de que sea respuesta a un agente infeccioso o tóxico y así
se han descrito cuadros semejantes a la esclerodermia como son el síndrome
del aceite tóxico, la enfermedad de Lyme o las reacciones a los
implantes cosméticos de silicona. La patofisiología de la
esclerodermia no está bien establecida existiendo en su desarrollo
factores vasculares, inflamatorios y especialmente el aumento de la síntesis
y deposito de colágeno en los tejidos afectos.
En relación a la intensidad
de la afectación cutánea y sistémica, se puede dividir
en dos grupos, esclerodermia sistémica limitada y difusa.
La forma sistémica limitada
tiene una esclerosis cutánea limitada, generalmente acral, afectando
a zonas distales de muñecas y tobillos, de curso más lento,
con afectación de órganos sistémicos menos evidente
y con fenómeno de Raynaud de largo tiempo de evolución (Tabla
XI), cuando la forma limitada se acompaña de calcinosis, raynaud,
alteraciones en la motilidad esofágica, esclerodactilia y telangiectasias,
se conoce con el acrónimo de síndrome de CREST.
La forma difusa se caracteriza
por el desarrollo de esclerosis cutánea generalizada por encima
de las muñecas, con afectación de órganos internos
y fenómeno de Raynaud de poco tiempo de evolución. En estos
pacientes la afectación vascular suele ser más importante.
|
Tabla XI
Formas clínicas de la esclerodermia sistémica
|
|
limitada
|
difusa
|
-
Fenómeno de Raymaud de larga evolución
-
Afectación cutánea limitada (partes acras)
-
Desarrollo tardío de telangiectasias, calcificaciones
y fibrosis pulmonar
-
Anticuerpos anticentrómero positivos
|
-
Intervalo corto entre el inicio del fenómeno de Raynaud
y el desarrollo de cambios cutáneos
-
Afectación troncal y periférica
-
roces de fricción tendinosos
-
fibrosis pulmonar, insuficiencia renal, afectación
digestiva y miocárdica
-
hemorragia capilar en pliegue ungueal
-
Ac Anti-SCL 70 positivos
-
Ac Anticentrómero negativos
|
La esclerodermia sistémica
se caracteriza por la existencia de dos marcadores cutáneos presentes
en la mayoría de pacientes, el fenómeno de Raynaud
y la acroesclerosis.
-
El fenómeno
de Raynaud se observa en el 98% de los pacientes con esclerodermia
y en el 70% es el síntoma inicial. Este fenómeno consiste
en episodios de isquemia digital que son desencadenados por factores como
el frio o las emociones. Típicamente se manifiesta por palidez en
los dedos, seguido de cianosis y rubor. La palidez indica el vasoespasmo,
la cianosis representa la isquemia debida a la disminución del oxígeno
en la sangre venosa y el rubor refleja la hiperemia reactiva tras el retorno
del flujo sanguíneo. El fenómeno de Raynaud asociado a esclerodermia
sistémica suele acompañarse de cambios capilares en la cutícula ungueal.
-
La acroesclerosis cutánea se manifiesta
inicialmente en las manos, que muestran edema y tumefacción de los
dedos, mientras que en los pliegues ungueales aparecen lesiones eritematosas,
siendo posible observar la existencia de capilares dilatados. Cuando existe
afectación de vasos de mayor calibre pueden desarrollarse infartos
en el pulpejo de uno o más dedos que darán lugar al desarrollo
de cicatrices y áreas atróficas. La fase más avanzada
de la enfermedad se caracteriza por una marcada esclerosis que puede acompañarse
de destrucción del tejido periungueal y óseo, y formación de
contracturas en flexión. Estos enfermos también pueden presentar afectación
facial. Los cambios más característicos
se observan alrededor de la boca, con aparición de fisuras y arrugas
y limitación en la apertura bucal (microstomia).
-
Los pacientes con esclerodermia
sistémica pueden desarrollar otros cambios cutáneos no relacionados
directamente con la esclerosis que incluirían calcinosis cutánea (nejm
2011),
alteraciones pigmentarias y cambios vasculares del tipo livedo reticularis
y vasculitis livedoide. La calcinosis suele observarse en los dedos de
la mano, es más frecuente en mujeres y en ocasiones sólo
es demostrable mediante estudios radiográficos.
Estudios serológicos
en pacientes con esclerodermia:
El 90% de los pacientes con
esclerodermia sistémica presentan auto-anticuerpos circulantes.
Los ANA suelen ser positivos en la mayoría de los pacientes. Existen
dos anticuerpos específicos de la esclerodermia que son el anti-centrómero
y el anti-Scl-70 (topoisomerasa I). Los anticuerpos anticentrómero
se observan en el 99% de los enfermos con la forma de esclerodermia
limitada o síndrome de CREST y su presencia en general confiere
un buen pronóstico. El Ac-anti-Scl-70 es altamente específico
de la forma difusa de esclerodermia sistémica su positividad se
relaciona con el desarrollo de fibrosis pulmonar y mal pronóstico.
El tratamiento de la esclerodermia
es difícil, los tratamientos están dirigidos a mejorar la
circulación, a reducir las reacciones inmunes involucradas en el
desarrollo de la esclerodermia y a reducir la síntesis de colágeno,
para lo cual se utilizan vasodilatadores, inmunosupersores y antifibróticos
(tabla XII). El tratamiento de la la esclerodermia localizada o morfea
incluye la aplicación tópica de corticoides, calcipotriol,
administración oral de calcitriol o la exposición a UVA y
PUVA.
|
Tabla XII
Tratamiento de la esclerodermia |
|
Localizada
|
Sistémica
|
-
Corticoides tópicos
-
Calcitriol y calcipotrieno
-
UVA y PUVA
-
Metotrexate
|
-
Vasodilatador
Nifedipina
Losartan
prostaciclinas y prostaglandinas
Bosentan
Sildenafilo
-
Inmunosupresor
Metotrexate
Ciclosporina
-
Antifibrótico
D-penicilamina
Fototerapia con UVA-1
|
Enfermedad del tejido conectivo
indiferenciada: Hasta un 50% de los pacientes con enfermedad del tejido
conectivo no pueden ser diagnosticados con un diagnóstico específico en los
primeros 12 meses de seguimiento. En estos casos se utiliza el término de
enfermedad del tejido conectivo indiferenciado. Algunos de estos pacientes
evolucionan hacia una enfermedad específica en los 5 años siguientes, un pequeño
grupo evolucionan de forma satisfactoria desapareciendo las manifestaciones , y
la mayoría de pacientes catalogados como enfermedad indiferenciada
presentan un curso benigno con manifestaciones sintomáticas pero sin tener
criterios de una enfermedad específica.
Enfermedad mixta del tejido
conectivo: la enfermedad mixta es un síndrome con hallazgos de esclerodermia
sistémica, lupus eritematoso sistémico y polimiositis asociado a la presencia de
anticuerpos anti U1RNP. Los hallazgos clínicos incluyen una alta frecuencia de
síndrome de Raynaud, manos edematosas, esclerodactilia, artritis, polimiositis y
enfermedad pulmonar interstiticial. Esta enfermedad suele tener un peor
pronóstico que el lupus sistémico, falleciendo los pacientes por hipertensión
pulmonar.
Síndromes
esclerodermiformes: Comprende un grupo amplio de enfermedades caracterizadas
por la esclerosis y endurecimiento cutáneo similar al observado en la
esclerodermia sistémica y morfea que pueden tener causas etiológicas
diversas que incluyen el depósito de mucina (escleromixedema), gamapatias
monoclonales, eosinofilia, diabetes, enfermedad injerto contra huesped, causas
químicas, hereditareas etc.
Nefropatía nefrogénica:
es una enfermedad fibrosante progresiva, que se observa en pacientes con
insuficiencia renal terminal que han sido tratados con hemodiálisis,
diálisis peritoneal o trasplante renal. La patogénesis de esta enfermedad no
está bien establecida, pero diversas observaciones relacionan su desarrollo con
la exposición agentes de contraste conteniendo galodinio utilizados en estudios
de imagen. El proceso fibrótico afecta a piel tejido subcutáneo, fascia, músculo
y también a órganos internos como pulmón y corazón.
Síndrome del aceite
tóxico:
Esta es una nueva enfermedad que apareció en
España como epidemia en 1981 relacionado con el consumo de aceite
denaturalizado con anilinas y comercializado fraudulentamente como aceite de
oliva. . Es una enfermedad multisistémica
que tiene varias fases de evolución. En la fase aguda (primer mes),
la enfermedad se caracteriza por fiebre, rash cutáneo, edema pulmonar
no cardiogénico y mialgia. La manifestación cutánea
más frecuente consiste en un eritema difuso con prurito. Histológicamente
muestra un edema dérmico difuso, con infiltración dérmica mononuclear, linfocítica y escasos eosinófilos. En las fases
más avanzadas de la enfermedad las lesiones son similares a la
esclerodermia.
Casos para dx en clase 20240305 podologia con asignación de alumnos
Casos
para dx en clase
test
patient page
www.uv.es/derma Dr. Víctor Alegre de
Miquel

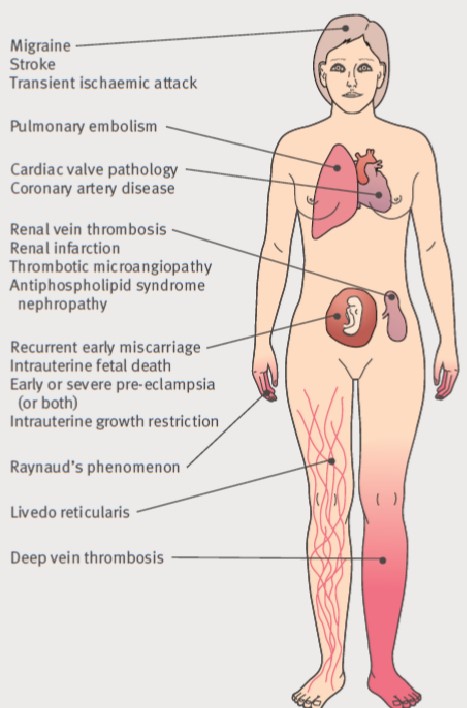 Síndrome de anticuerpos
antifosfolípidos: Es un síndrome caracterizado
por el desarrollo de trombosis venosas profundas, trombosis arteriales,
tombocitopenia y abortos de repetición asociado a la presencia de anticuerpos
anticardiolipina, anticoagulante lúpico o anticuerpos anti
β2-glicoproteina
I. El síndrome
de anticuerpos antifosfolípidos puede ser primario (>50% casos) o secundario
según esté asociado a otras enfermedades del tejido conectivo,
siendo la más frecuente el lupus eritematoso sistémico (entre el 20 y el 35% de
pacientes con lupus sistémico desarrollaran un síndrome de anticuerpos
antifosfolipídico secundario). Existe una variante
-Síndrome
antifosfolípidico catastrófico-
que da lugar a una microangiopatía trombótica diseminada y fallo multiorgánico. Las lesiones
cutáneas observadas en pacientes con síndrome de anticuerpos
antifosfolípidos incluyen el desarrollo de úlceras cutáneas, livedo reticularis, vasculitis livedoide y tromboflebitis superficial.
El estudio histológico de las lesiones cutáneas muestra la
presencia de una trombosis no inflamatoria de los vasos dérmicos.
Los anticuerpos antifosfolípidos pueden detectarse analíticamente
de 3 maneras: 1) anticuerpos
anticardiolipina, 2)Anticoagulante lúpico en plasma en dos o más ocasiones y 3)
anticuerpos anti
β2-glicoproteina
I, presentes en más de 2 ocasiones.; la medición
de los anticuerpos anticardiolipina es el test más sensible para
detectar su presencia, pero pueden ser positivos en otras enfermedades
autoinmunes, infecciones e incluso en la población normal.
Síndrome de anticuerpos
antifosfolípidos: Es un síndrome caracterizado
por el desarrollo de trombosis venosas profundas, trombosis arteriales,
tombocitopenia y abortos de repetición asociado a la presencia de anticuerpos
anticardiolipina, anticoagulante lúpico o anticuerpos anti
β2-glicoproteina
I. El síndrome
de anticuerpos antifosfolípidos puede ser primario (>50% casos) o secundario
según esté asociado a otras enfermedades del tejido conectivo,
siendo la más frecuente el lupus eritematoso sistémico (entre el 20 y el 35% de
pacientes con lupus sistémico desarrollaran un síndrome de anticuerpos
antifosfolipídico secundario). Existe una variante
-Síndrome
antifosfolípidico catastrófico-
que da lugar a una microangiopatía trombótica diseminada y fallo multiorgánico. Las lesiones
cutáneas observadas en pacientes con síndrome de anticuerpos
antifosfolípidos incluyen el desarrollo de úlceras cutáneas, livedo reticularis, vasculitis livedoide y tromboflebitis superficial.
El estudio histológico de las lesiones cutáneas muestra la
presencia de una trombosis no inflamatoria de los vasos dérmicos.
Los anticuerpos antifosfolípidos pueden detectarse analíticamente
de 3 maneras: 1) anticuerpos
anticardiolipina, 2)Anticoagulante lúpico en plasma en dos o más ocasiones y 3)
anticuerpos anti
β2-glicoproteina
I, presentes en más de 2 ocasiones.; la medición
de los anticuerpos anticardiolipina es el test más sensible para
detectar su presencia, pero pueden ser positivos en otras enfermedades
autoinmunes, infecciones e incluso en la población normal.