| HOME | HELP | FEEDBACK | SUBSCRIPTIONS | ARCHIVE | SEARCH | TABLE OF CONTENTS |
|
| ||||||||
J. Biol. Chem., Vol. 280, Issue 8, 6950-6959, February 25,
2005
 -Glutamyltranspeptidase Sensitize Metastatic B16 Melanoma Cells to
Endothelium-induced Cytotoxicity*
-Glutamyltranspeptidase Sensitize Metastatic B16 Melanoma Cells to
Endothelium-induced Cytotoxicity*
 ¶**
¶**From the Departamento de Fisiología, Universidad de Valencia, 46010
Valencia, Spain and the ||Centro Nacional de Investigaciones
Oncológicas, 28029 Madrid, Spain
Received for publication, July 28, 2004 , and in revised form, November 19, 2004.
| ABSTRACT |
|---|
.gif) -glutamyltranspeptidase (which limits GSH synthesis by
preventing cysteine generation from extracellular GSH). When applied
under in vivo conditions, this strategy increased tumor
cytotoxicity (up to
-glutamyltranspeptidase (which limits GSH synthesis by
preventing cysteine generation from extracellular GSH). When applied
under in vivo conditions, this strategy increased tumor
cytotoxicity (up to  90%)
during B16M-F10 cell adhesion to the hepatic sinusoidal
endothelium.
90%)
during B16M-F10 cell adhesion to the hepatic sinusoidal
endothelium.
| INTRODUCTION |
|---|
-glutamylcysteinylglycine) content show higher metastatic
activity in the liver than those with low GSH content (1). The liver is
a common site for metastasis development, and we demonstrated that
GSH protects B16M cells against nitrosative and oxidative stress in
the murine hepatic microvasculature (2, 3). The concept
that high GSH content status is an important factor for metastasis
progression was strongly supported by the fact that metastatic B16M
cell survival and growth can be enhanced by directly increasing their
GSH content with GSH ester, which readily enters the cell and
delivers free GSH (4, 5). In
consequence, the maintenance of high intracellular levels of GSH
appears critical for metastatic cells to survive intravascularly and
to progress extravascularly.
Multidrug and/or radiation resistance, which are characteristic
features of malignant tumors, frequently associate with high
GSH content in the cancer cells (6). Efflux of GSH
and GSH S-conjugates from different mammalian cells is
mediated by multidrug resistance proteins (MRP), among which MRP1 and
MRP2 have been characterized at the functional level as ATP-dependent
pumps with broad specificity for GSH and glucuronic or sulfate
conjugates (7–10).
Multidrug resistance frequently associates with the overexpression
of P-glycoprotein and/or MRP1 (11), both
functioning as pumps that extrude drugs from tumor cells. GSH
depletion induced by L-buthionine-(SR)-sulfoximine (BSO), a specific
inhibitor of -glutamylcysteine synthetase (the rate-limiting step in GSH
biosynthesis) (12),
resulted in a complete reversal of resistance to anticancer drugs
of different cell lines overexpressing MRP1 but had no effect on
P-glycoprotein-mediated multidrug resistance (13). Most
interestingly, cancer cells can release GSH through MRP1 even in the
absence of cytotoxic drugs (7).
In a recent study we demonstrated that B16M-F10 cells with a high
metastatic potential overexpress BCL-2, show an increase in
intracellular GSH content, show no change in the GSH synthesis rate,
but show a decrease in GSH efflux (5). This study
also provides evidence that BCL-2 can directly inhibit GSH
export, thereby accounting for the increase in intracellular GSH.
Moreover, it demonstrates that GSH depletion and BCL-2 antisense
therapy can sensitize cells to TNF- -induced
apoptotic death. In consequence, it is plausible that BCL-2, in
addition of its anti-apoptotic properties (14), may also
increase metastatic cell resistance against oxidative/nitrosative
stress by preserving intracellular GSH. In fact, GSH efflux prior or
during apoptosis appears an essential regulator of the apoptotic
killing mechanism (15).
-induced
apoptotic death. In consequence, it is plausible that BCL-2, in
addition of its anti-apoptotic properties (14), may also
increase metastatic cell resistance against oxidative/nitrosative
stress by preserving intracellular GSH. In fact, GSH efflux prior or
during apoptosis appears an essential regulator of the apoptotic
killing mechanism (15).
Because GSH efflux from B16M cells can be increased by using Bcl-2 antisense oligodeoxynucleotides (Bcl-2-AS) (5), and possibly by using different MRP1 regulators, the aim of the present report was to investigate whether the rate of efflux may become an important factor regulating intracellular GSH content and thereby metastatic cell survival.
| EXPERIMENTAL PROCEDURES |
|---|
GSH and GSSG—GSH was measured by the glutathione S-transferase reaction and GSSG by high pressure liquid chromatography (4). Total glutathione (GSH + 2GSSG) was determined by a kinetic assay in which a catalytic amount of GSH or GSSG and glutathione reductase cause the continuous reduction of 5,5'-dithiobis-2-nitrobenzoic acid (Sigma) by NADPH (16). GSH monoisopropyl(glycyl) ester was prepared as described previously (17).
Bcl-2 Gene Transfer and BCL-2 Analysis—The Tet-Off gene expression system (Clontech) was used, as in Ref. 5, to insert the mouse Bcl-2 gene and for transfection into B16M cells following the manufacturer's instructions. BCL-2 protein was quantitated in the soluble cytosolic fraction by enzyme immunoassay (18) using a monoclonal antibody-based assay from Sigma (1 unit of BCL-2 was defined as the amount of BCL-2 protein in 1,000 nontransfected B16M cells).
Cellular BCL-2 protein levels were also analyzed, as recently
reported (5), by flow
cytometry using an EPICS PROFILE II (Coulter Electronics, Hialeah,
FL), at 488 nm and 250 milliwatts. Cellular suspensions were diluted
to 250,000 cells/ml. Primary monoclonal anti-BCL-2 from mouse
followed by biotin-conjugated goat anti-mouse IgG and
phycoerythrin-labeled streptavidin (Sigma) were used. BCL-2 protein
levels were expressed as arbitrary fluorescence units ( FL1). Cell viability in these experiments was determined
with propidium iodide (final concentration 10 µM,
Molecular Probes, Eugene, OR).
FL1). Cell viability in these experiments was determined
with propidium iodide (final concentration 10 µM,
Molecular Probes, Eugene, OR).
Electroporation and Selection of B16M-F10 Multidrug Resistance
Protein Knock-out Clones—Murine MRP genomic sequences were
isolated from a 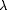 Charon
35 phage library constructed with DNA from mouse strain C57BL/6J. The
preparation of the gene-targeting construct, electroporation into
tumor cells (Bio-Rad), and selection and screening of the clones,
first by PCR analysis and then by DNA blot analysis, were carried out
as described previously (19). The
multidrug resistance associated protein-1 or -2 knock-out clones
(MRP-/-1 and MRP-/-2) were obtained by exposing a single knock-out
clone to high concentrations of G418 for 2 weeks.
Charon
35 phage library constructed with DNA from mouse strain C57BL/6J. The
preparation of the gene-targeting construct, electroporation into
tumor cells (Bio-Rad), and selection and screening of the clones,
first by PCR analysis and then by DNA blot analysis, were carried out
as described previously (19). The
multidrug resistance associated protein-1 or -2 knock-out clones
(MRP-/-1 and MRP-/-2) were obtained by exposing a single knock-out
clone to high concentrations of G418 for 2 weeks.
Treatment of B16M-F10 Cells with Oligodeoxynucleotides—B16M-F10 cells were incubated, as described previously (5), in the presence of Bcl-2 antisense phosphorothioate-derivatized oligodeoxynucleotides (Bcl-2-AS) from Eurobio (Les Ulis, France). Fresh oligodeoxynucleotides were added every day, starting 24 h after cell plating, as 1:1 complexes with the Lipofectin transfection reagent (Invitrogen). The sequence of Bcl-2-AS corresponding to the mouse Bcl-2 translation initiation codon site and extending 3' downstream for a total of 18 bases was 5'-TCTCCCGGCTTGCGCCAT-3' (20). A 2-base Bcl-2 mismatch oligodeoxynucleotide (5'-TCTCCCGGCATGTGCCAT-3') was used as control.
Quantitative Determination of the Plasma Membrane Potential— Plasma membrane potential (PMP) was measured using a standard technique (21). B16M-F10 cells were cultured for 24 h, as described above, but seeded at 5 x 104 cells/cm2 in glass Petri dishes. Culture dishes were mounted on a tube-focusing microscope (Nikon, Tokyo, Japan). Intracellular measurements were performed, at 25 °C, with glass micropipettes filled with 3 M KCl and with a 20-megohm DC resistance. Membrane potentials were measured with a WP Instruments M4-A electrometer amplifier (Sarasota, FL), and the output was displayed using a MacLab System (Castle Hill, Australia). Measurements were made only in cells (from at least four different preparations) that gave a stable membrane potential within 10 s of penetration, which indicates a good seal of the plasma membrane with the recording electrode.
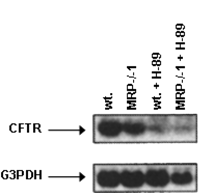
Western Blots—Cultured cells were harvested, as reported previously (1), and then washed twice in ice-cold Krebs-Henseleit bicarbonate medium, pH 7.4. Whole cell extracts were made by freeze-thaw cycles in a buffer containing 150 mM NaCl, 1 mM EDTA, 10 mM Tris-HCl, 1 mM phenylmethylsulfonyl fluoride, 1 µg/ml leupeptin, 1 µg/ml aprotinin, and 1 µg/ml pepstatin, pH 7.4. Fifty µg of protein (as determined by the Bradford assay) were boiled with Laemmli buffer and resolved in 12.5% SDS-PAGE. Proteins were transferred to a nitrocellulose membrane and subjected to Western blotting with anti-cystic fibrosis transmembrane conductance regulator (anti-CFTR) monoclonal antibody (CFTR (CF3) antibody from Novus Biologicals, Littleton, CO). Immunizing peptide corresponds to amino acid residues 103–1117 found in the first extracellular loop of human CFTR. This sequence is conserved in mice. Blots were developed using horseradish peroxidase-conjugated secondary antibody and enhanced chemiluminescence (ECL system, Amersham Biosciences).
Reverse Transcription-PCR—A 392-bp region corresponding to nucleotides 1340–1730 of the mouse CFTR gene (22) was amplified with the forward primer 5'-GGGAGGAGGGATTTGGGGAA-3' and the reverse primer 5'-GTGATGTCCTGCTGTAGTTG-3'. The CFTR cDNA was obtained using a random hexamer primer and a MultiScribe Reverse Transcriptase kit as described by the manufacturer (TaqMan RT Reagents, Applied Biosystems, Foster City, CA). A PCR master mix containing the specific primers and AmpliTaq Gold DNA polymerase (Applied Biosystems) were then added. Amplifications were performed in a GeneAmp 2400 thermal cycler (PerkinElmer Life Sciences).
Northern Blot Analysis—Total RNA was isolated using the TRIzol kit from Invitrogen and following the manufacturer's instructions. Ten µg of total RNA were electrophoresed in 1% agarose gels containing formaldehyde. RNA was electrophoretically transferred to a nylon membrane (GeneScreen; PerkinElmer Life Sciences) and covalently bound to the membrane following UV cross-linking. Murine CFTR and control glyceraldehyde-3-phosphate dehydrogenase (G3PDH, Clontech) cDNA probes were radiolabeled with 32P by random priming (23). The membranes were hybridized at 65 °C with the 32P-labeled cDNA fragments. After washing at room temperature, the filters were exposed to film, and autoradiography was performed using a PhosphorImager (Bio-Rad).
Perifusion of B16M-F10 Cells—Isolated B16M cells, suspended in DMEM, were incubated in a perifusion system similar to that described previously for rat hepatocytes (24). Briefly, a buffer gassed with 95% O2, 5% CO2 was constantly pumped by an LKB multiperpex roller pump (type 2115; Amersham Biosciences) to a chamber containing a final volume of 10 ml and 3 x 106 B16M-F10 cells per ml. The filter (Ultracel Amicon YM100 membrane, Millipore, Billerica, MA) was placed at the top of the chamber. The tumor cell suspension was perifused at 37 °C and maintained at a homogenous state by using a magnetic stirrer placed at the bottom of the chamber. The perifusion buffer was Krebs-Henseleit bicarbonate medium, pH 7.4, containing plasma concentrations (aortic blood) of free L-amino acids found in tumor-bearing C57BL/6J mice (395 ± 34 µM Ala, 56 ± 8 µM Asn, 24 ± 3 µM Asp, 8 ± 1 µM Cyst(e)ine, 72 ± 18 µM Glu, 442 ± 37 µM Gln, 229 ± 36 µM Gly, 46 ± 11 µM His, 62 ± 13 µM Ile, 78 ± 16 µM Leu, 265 ± 23 µM Lys, 44 ± 6 µM Met, 56 ± 9 µM Phe, 107 ± 11 µM Pro, 87 ± 24 µM Ser, 117 ± 14 µM Thr, 70 ± 15 µM Trp, 83 ± 10 µM Tyr, 115 ± 18 µM Val, 129 ± 16 µM Arg; n = 15), GSH (9 ± 1 µM), glucose (1 g/liter), sodium pyruvate (10 mg/liter), albumin-free fetal calf serum (1.0%, Invitrogen) and supplemented with 10 units/ml penicillin and 10 µg/ml streptomycin. Perfusate flow (2 ml/min) was constant throughout the experiment. Effluent flow was monitored continuously for O2 and pH with Philips electrodes. Tumor cell viability was always >97% along the experimental time. A syringe was introduced into the chamber through a rubber septum to take samples (0.5 ml) of the cell suspension without interrupting the flow.
Amino Acid Analysis—Proteins were precipitated by treating 0.1 ml of intracellular compartment or plasma with 0.4 ml of 3.75% (w/v) ice-cold sulfosalicylic acid in 0.3 M lithium citrate buffer, pH 2.8. After centrifugation, 0.25 ml of the supernatant were injected into an LKB 4151 amino acid analyzer (Amersham Biosciences) (25).
Verapamil and Acivicin Cellular Pharmacokinetics—Accumulation
of N-methyl[3H]verapamil (VRP) (-[3-[[2-(3,4-dimethoxyphenyl)ethyl]
methylamino]propyl]-3,4-dimethoxy--(1-methylethyl)monohydrochloride) or
[3H]acivicin (ACV) (L-(S,5S)--amino-3-chloro-4-dihydro-5-isoxazole acetic acid)
(PerkinElmer Life Sciences) in perifused cells was measured as
described previously (26, 27). Briefly,
B16M-F10 cells were perifused (see above) in the presence of 1 µM [3H]VRP (0.05 µCi/ml) or 2 µM [3H]ACV (0.1 µCi/ml). Aliquots of
perifused cells (0.2 ml) were taken from the chamber each 10 min, and
accumulation of [3H]VRP or [3H]ACV was stopped
by rapid dilution into ice-cold PBS. Cells were centrifuged and
washed twice with 1 ml of ice-cold PBS. After centrifugation, cell
pellets were solubilized in 1% SDS, and cell-associated radioactivity
was determined by liquid scintillation counting.
GSH-related Enzyme Activities—B16M-F10 cells were detached
(see above), washed twice at 4 °C in Krebs-Henseleit bicarbonate
medium (without Ca2+ or Mg2+) containing 0.5 mM EGTA, pH 7.4, resuspended, and homogenized in 0.1
M phosphate buffer, pH 7.2, at 4 °C. -Glutamylcysteine synthetase (-GCS) and GSH synthetase activities were measured as
described elsewhere (4).
-Glutamylcysteine Synthetase Expression Analysis—Total
RNA was isolated by the acid phenol guanidine method (28). cDNA
was obtained using a random hexamer primer and a MultiScribe
reverse transcriptase kit as described by the manufacturer (TaqMan
Reverse Transcription Reagents, Applied Biosystems, Foster City,
CA). A PCR master mix containing the specific primers (-GCS-heavy subunit (-GCS-HS): forward, 5'-ATC CTC CAG TTC CTG CAC ATC TAC, and
reverse 5'-GAT CGA AGG ACA CCA ACA TGT ACTC; -GCS-light subunit (-GCS-LS): forward, TGG AGT TGC ACA GCT GGA CTC T, and
reverse 5'-CCA GTA AGG CTG TAA ATG CTC CAA; G3PDH: forward,
5'-CCT GGA GAA ACC TGC CAA GTA TG, and reverse 5'-GGT CCT CAG
TGT AGC CCA AGA TG) and AmpliTaq Gold DNA polymerase (Applied
Biosystems) were then added. Real time quantitation of -GCS-HS and -GCS-LS mRNA relative to G3PDH mRNA was performed with a
SYBR Green I assay and evaluated using a iCycler detection system
(Bio-Rad). Target cDNAs were amplified in separate tubes using
the following procedure: 10 min at 95 °C and then 40 cycles of
amplification (denaturation at 95 °C for 30 s and annealing and
extension at 60 °C for 1 min per cycle). The increase in fluorescence
was measured in real time during the extension step. The threshold
cycle (CT) was determined, and then the relative
gene expression was expressed as follows: fold change = 2-(CT), where CT = CT target -
CT G3PDH, and (CT) = CT treated - CT control.
Measurement of H2O2—The assay of H2O2 production was based on the H2O2-dependent oxidation of the homovanillic acid (3-methoxy-4-hydroxyphenylacetic acid) to a highly fluorescent dimer (2,2'-dihydroxydiphenyl-5,5'-diacetic acid) that is mediated by horseradish peroxidase (3).
Cystine Uptake—B16M-F10 cells were plated in 25-cm2 culture dishes. At the required times, cells were rinsed three times with prewarmed transport medium (10 mM PBS, pH 7.4, with 0.01% CaCl2, 0.01% MgCl2 and 0.1% glucose). Uptake measurement was initiated by addition of 1.0 ml of transport medium containing 1 µCi of L-[3H]cystine (Amersham Biosciences) and nonradioactive L-cystine (0.5 mM). After incubation at 37 °C, uptake was finished by rinsing several times with ice-cold PBS until less than 0.001% of the initial radioactivity was present in the supernatant. Cells were then dissolved with 0.5 ml of 0.5 N NaOH, and an aliquot was used for determining radioactivity and another for protein assay. To correct for trapping, transport at 4 °C was studied in parallel (29).
-Glutamyltranspeptidase Assay—-GT activity was measured as described previously (30). Protein was
determined with the BCA protein assay (Pierce).
Isolation and Culture of Hepatic Sinusoidal Endothelium—Syngenic male C57BL/6J mice (10–12 weeks old) were from IFFA Credo (L'Arbreole, France) and received care according to the criteria outlined by the National Institutes of Health. Hepatic sinusoidal endothelium (HSE) was separated in a 17.5% (w/v) metrizamide gradient and identified as described previously (3). Cultures of HSE were established and maintained in pyrogen-free DMEM supplemented as described above for the B16M cells. Differential adhesion of endothelial cells to the collagen matrix and washing allows a complete elimination of other sinusoidal cell types (Kupffer, stellate, lymphocytes) from the culture flasks.
B16M-F10-Endothelial Cell Adhesion and Cytotoxicity Assays— B16M-F10 cells were loaded with 2',7'-bis(2-carboxyethyl)-5,6-carboxyfluorescein acetoxymethyl ester (BCECF-AM, Molecular Probes) (106 cells were incubated in 1 ml of HEPES buffered DMEM, containing 50 µg of BCECF-AM and 5 µl of Me2SO, for 20 min at 37 °C). After washing, BCECF-AM-containing cells were resuspended in HEPES buffered DMEM without phenol red at a concentration of 2.5 x 106 cells/ml and added (0.2 ml/well) to endothelial cells (plated 24 h before) and also to plastic or collagen pre-coated control wells. The plates were then incubated at 37 °C, and 20 min later the wells were washed three times with fresh medium and read for fluorescence using a Fluoroskan Ascent FL (Labsystems, Manchester, UK). The number of adhering tumor cells was quantified by arbitrary fluorescence units based on the percentage of the initial number of B16M-F10 cells added to the HSE culture (3). Damage to B16M-F10 cells during their in vitro adhesion to the HSE was measured, as described previously (2), using tumor cells loaded with calcein-AM (Molecular Probes). The integrity of B16M-F10 cells cultured alone was assessed by trypan blue exclusion and by measuring lactate dehydrogenase activity released to the extracellular medium (1). Other reagents used in experiments of tumor cytotoxicity were from Sigma.
Cytokines—Recombinant murine TNF- (2 x 107 units/mg protein) and
recombinant murine (IFN-; 105 units/mg protein) were obtained from Sigma.
Stock solutions (5 x 105 units
TNF-/ml and
25 x 104 units of
IFN-/ml) were diluted in sterile physiological saline solution
(0.9% NaCl), adjusted to pH 7.0, and stored at 4 °C.
In Vivo Microscopy—C57BL/6J mice were fed ad libitum on a stock laboratory diet (Letica, Barcelona, Spain) and kept on a 12-h light/12-h dark cycle with the room temperature maintained at 22 °C. Procedures involving animals were in compliance with the national and international laws and policies (EEC Directive 86/609, OJ L 358. 1, December 12, 1987; and National Institutes of Health Guide for the Care and Use of Laboratory Animals Number 85-23). In vivo microscopy to follow metastatic cell dynamics within the liver was performed, as earlier described (2), using calcein-AM (Molecular Probes)-labeled B16M-F10 cells. The total number of calcein-AM-labeled cells per hepatic lobule was recorded in 10 different lobules per liver at 15-min intervals for a 6-h period. The microscope was an Eclipse E600FN, providing transillumination or epi-illumination, equipped for video microscopy using a digital DXM 1200 camera (Nikon, Tokyo, Japan).
Apoptosis Detection—The livers used for in vivo microscopy studies were fixed in formalin embedded in paraffin and then processed in 10-µm thick sections for routine hematoxylin staining. Immunohistochemical detection of apoptosis at single cell level, based on labeling of DNA strand breaks (TUNEL), was followed using the in situ cell death detection kit POD from Roche Diagnostics. Sections were examined under a Leica Microsystems (Nussloch, Germany) upright microscope with standard x10, x40, and x63 plan apo-objectives.
Experimental Metastases—Hepatic metastases were produced by intravenous injection (portal vein) into anesthetized mice (nembutal, 50 mg/kg intraperitoneally) of 4 x 105 viable B16M-F10 cells suspended in 0.2 ml of DMEM. Mice were cervically dislocated 10 days after B16M-F10 inoculation. The livers were fixed with 10% formaldehyde in PBS (pH 7.4) for 24 h at 22 °C and then paraffin-embedded. Metastasis density (mean number of foci/100-mm3 of liver detected in 15 10 x 10-mm2 sections per liver) and metastasis volume (mean % of liver volume occupied by metastasis) were determined as described earlier (1).
Expression of Results and Statistical Significance—Data were analyzed by one- or two-way ANOVA or t tests where appropriate (SPSS 9.0 software for Windows; SPSS Inc., Chicago). The homogeneity of the variances was analyzed by the Levene test. The null hypothesis was accepted for all the values of the tests in which the F value was nonsignificant at p > 0.05. The data for which the F value was significant was examined by Tukey's test at p < 0.05.
| RESULTS |
|---|
We investigated further the mechanisms through which GSH is released from highly metastatic cells. As shown in Table I, GSH efflux from B16M-F10 cells was decreased by L-methionine suggesting that, in part, GSH is released through a system possibly similar to an L-methionine-sensitive sinusoidal GSH transporter detected in hepatocytes (31, 32). However, GSH efflux was not inhibited by L-methionine in B16M-F10/Tet-BCL-2 (Table I), suggesting that likely BCL-2 and L-methionine act on the same transport system (similar results were reported previously in HeLa cells (33)). Further experiments using MRP-/-1 or MRP-/-2 clones showed that GSH efflux was decreased, as compared with controls, in MRP-/-1 B16M-F10 and in B16M-F10/Tet-BCL-2 cells (Table I). GSH efflux in MRP-/-1 B16M-F10/Tet-BCL-2 cells was practically abolished, and identical results were obtained in MRP-/-1 B16M-F10 cells incubated in the presence of methionine (Table I). Taken together these results indicate that a BCL-2-dependent system and MRP1 are the main mechanisms channeling GSH efflux from B16M-F10 cells.
|
63% of
control values (see above) but did not affect significantly rates
of GSH efflux measured in controls, in the presence of L-methionine (1 mM) or in MRP-/-1
cells (see e.g. Table I).
Apparently, efflux of GSH is not driven in B16M-F10 cells by a
significant change in the PMP. Our results appear in agreement with
those reported by Liu et al. (37) for Hep G2
cells, a human-derived hepatoma cell line, where a fall in the
membrane potential produced by the replacement of Na+ with
equivalent K+ did not affect GSH efflux significantly.
Neither ouabain, vanadate (a Ca2+-ATPase inhibitor), nor
BaCl2 (a K+ channel blocker) significantly affected
the GSH efflux; however, L-methionine (1 mM) decreased GSH efflux from Hep G2 cells (37).
|
3% of
control BCL-2 levels, see the legend to Fig. 1), whereas
GSH efflux decreased to 50% of
control values in MRP-/-1 B16M-F10 cells (Fig. 1). The
difference represents the BCL-2-dependent GSH efflux (see also data
in Table I).
Monoclonal antibodies anti-CFTR practically abolished the
BCL-2-dependent GSH efflux from B16M-F10 cells (Fig. 1; the
presence of CFTR in B16M-F10 cells was confirmed with a Western blot,
not shown). In consequence, addition of anti-CFTR antibodies to
Bcl-2-AS-treated MRP-/-1 B16M-F10 cells practically abolished
GSH efflux (Fig.
1).
|
|
Antisense Bcl-2 Oligodeoxynucleotides and Verapamil Accelerate
GSH Release from B16M-F10 Cells—Parallel to the BCL-2-sensitive
CFTR, two mechanisms of transport of GSH by MRP1 have been suggested
as follows: passive permeability and a VRP-dependent active
transport (42). VRP, an
inhibitor of P-glycoprotein-mediated drug efflux, is not transported
by MRP1 (43)
but may also inhibit MRP1-mediated drug extrusion (44). We tested
VRP in combination with Bcl-2-AS treatment to potentiate GSH
efflux from B16M-F10 cells. A perifusion chamber, containing a
suspension of B16M-F10 cells, was used as an experimental setup that
mimics in vivo conditions by providing a constant supply of
glucose, amino acids, and GSH at physiological plasma concentrations
(see under "Experimental Procedures"). This setup allowed us to use a
VRP concentration (1 µM) that corresponds to
clinically accepted and nontoxic levels in plasma (45)
(dose-response studies showed that 0.5–2 µM VRP
activates GSH efflux from perifused cells in a
concentration-dependent fashion, data not shown). VRP accumulation
within perifused B16M-F10 cells peaked 10–20 min (70
pmol/106 cells) after addition to the perfusate flow and
then decreased reaching a lower steady-state concentration at 60 min
(40 pmol/106 cells) (Fig. 3). These
values of VRP accumulation, which are in agreement with those
reported previously in HeLa cells (43), were not
changed significantly when control and BCL-2-AS-treated cells were
compared (Fig.
3).
|
100%, Table II).
Intracellular GSH contents were significantly decreased (30%), as
compared with controls, in Bcl-2-AS- and VRP-treated B16M-F10
cells after 6 h of perifusion (Table III).
However, at 12 h of perifusion time, GSH levels were 70% higher
in Bcl-2-AS- and VRP-treated B16M-F10 cells than in controls
(Table II).
Thus, it appeared plausible that loss of GSH accelerates their rate
of intracellular synthesis.
|
-Glutamylcysteine Synthetase Overexpression in B16M-F10
Cells—As shown in Table III, either
both or Bcl-2-AS or VRP treatment significantly increased the
rate of GSH synthesis in B16M-F10 cells. Moreover, their effects
appeared additive and associated with an increase in -GCS activity (Table III).
Bcl-2-AS and/or VRP, as compared with controls, did not change
significantly the GSH synthetase activity (see Table III
legend). Moreover, as shown in Table IV,
the increase in -GCS activity was accompanied by a previous increase in both
-GCS-HS and -GCS-LS expression (maximum values were found at 3 h).
Therefore, increased GSH efflux associates with -GCS overexpression.
|
-GCS activity can result from transcriptional and
post-transcriptional regulation affecting the HS and/or the LS
(for review see Ref. 46).
Intracellular GSH depletion, e.g. that induced by
GSH-conjugating agents such as diethyl maleate, increased
transcription of both subunits (46) as it occurs
in our experimental conditions. Moreover, oxidative stress,
which may arise as a consequence of GSH depletion and increased
intracellular levels of H2O2 and OH·, can also
induce increased transcription of both subunits (46). In fact,
Bcl-2-AS- and VRP-induced GSH depletion (Table II) is
accompanied by an increase in H2O2 production
by the B16M-F10 cells (13 ± 4 in controls and 25 ± 5 nmol of
H2O2/106cells x h in Bcl-2-AS- and VRP-treated cells,
n = 5, p < 0.01).
Inhibition of -Glutamyltranspeptidase Activity Prevents GSH Content
Increase in B16M-F10 Cells Treated with Antisense Bcl-2
Oligodeoxynucleotide and Verapamil—In rapidly growing
tumors cyst(e)ine, whose concentration in blood is low, may
become limiting for GSH synthesis and cell growth (4, 47). Thus,
in order to increase the rate of GSH synthesis, malignant cells
may require alternative pathways to ensure cyst(e)ine availability.
Keeping the extracellular supply of amino acids constant and
physiological during the perifusion (see under "Experimental
Procedures"), the intracellular availability of amino acid precursors
for GSH synthesis was investigated. The concentrations of free
L-glutamate and glycine within the B16M-F10 cells
were constant through the perifusion time (e.g. 3.3 ± 0.4 and
2.5 ± 0.3 mM, respectively, in controls;
n = 6; enough to ensure maximum rates of GSH synthesis (12)) and were
not changed by addition of Bcl-2-AS and/or VRP (not shown).
However, free intracellular L-glutamine and
L-cyst(e)ine were undetectable, which is not
surprising because L-glutamine is a major fuel used
by cancer cells (48), and L-cyst(e)ine is rapidly used for protein and GSH
synthesis (25). L-Cystine is predominant outside the cell because
L-cysteine rapidly autoxidizes to L-cystine in the extracellular fluids, but once it
enters the cell through the Xc- system L-cystine is reduced to L-cysteine (Ref. 46 and
references therein). Thus, we measured L-cystine
uptake by B16M-F10 cells and found that Bcl-2-AS and/or VRP
did not significantly affect this rate (e.g. 0.41 ± 0.08
nmol/mg protein in controls, n = 5). Nevertheless, we showed
that tumor -GT activity and an intertissue flow of GSH increase GSH
content in B16M-F10 cells and work as a tumor growth-promoting
mechanism (4).
-GT cleaves extracellular GSH-releasing -glutamyl-amino acids and cysteinylglycine, which is further
cleaved by membrane-bound dipeptidases into L-cysteine and glycine (12). Free -glutamyl amino acids, L-cysteine and glycine,
entering the cell serve as GSH precursors (12). Hence, -GT expression may provide tumor cells with a growth
advantage at physiological concentrations of L-cyst(e)ine (4, 47). Therefore,
if the increase in GSH content (Table II)
following the effect of Bcl-2-AS and VRP on GSH efflux depends
on L-cyst(e)ine availability, and if this is
provided in part by the -GT, then inhibition of this activity could limit GSH
synthesis. We tested this possibility by adding to the perifusion
system ACV, an irreversible -GT inhibitor (49). ACV was
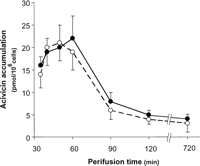
present in the perfusate flow for only 30 min (Fig. 4). ACV
accumulation within the B16M-F10 cells peaked 10–20 min (20
pmol/106 cells) after addition, and then its concentration
decreased to a steady-state low level (4
pmol/106 cells) (Fig. 4; these
values were not changed when control and Bcl-2-AS- or
VRP-treated cells were compared, data not shown). ACV decreased -GT activity to nondetectable levels and decreased GSH
synthesis but did not affect the rate of GSH efflux (Table V) or the
rate of L-cystine uptake (not shown, see above
for control values). However, the rate of GSH synthesis in B16M-F10
cells treated with Bcl-2-AS, VRP, and ACV was found similar to
controls (as under "no additions" in Table V)
when the concentration of L-cysteine in the
perifusion buffer was increased 2-fold (up to 16 µM). Hence, intracellular L-cysteine
availability indeed appears modulated by its -GT-dependent generation from extracellular GSH. Therefore,
we conclude that Bcl-2-AS- and VRP-induced acceleration of GSH
efflux combined with inhibition of -GT promote GSH depletion in B16M-F10 cells.
|
|
96%) than
in the presence of BSO (66%) (Table VI).
This is in agreement with our previous report (5) showing
that depletion of GSH and BCL-2 levels practically avoids
B16M-F10 survival during interaction with the vascular endothelium.
Also in agreement with this previous report (5), GSH depletion
did not alter the % of tumor cell adhesion to the HSE. To prove
that GSH is directly regulating metastatic cell survival, we
tested the effect of GSH ester (which enters the cell and delivers
free GSH (4)). As shown in
Table VI, GSH
ester prevented HSE-induced B16M-F10 cytotoxicity in the presence of
BSO, but only partially in the presence of Bcl-2-AS, VRP, and
ACV (where BCL-2 depletion remains, see Ref. 5). Second, we
assayed the effect of Bcl-2-AS, VRP, and ACV on the in
vivo B16M-F10 cell arrest and viability within the hepatic
microvasculature. Calcein-labeled B16M-F10 cells, which present a
green fluorescent cytoplasm, arrested in the liver sinusoids after
intraportal inoculation (Table VII).
The number of arrested B16M-F10 and Bcl-2-AS-treated B16M-F10
cells was similar (Table VII) and
constant along the time (no significant differences were found within
the 30–360-min postinjection) (not shown). As reported previously (2), the
number of arrested cells was not altered by depleting tumor
cell GSH or by treating mice with lipopolysaccharide (LPS) (to
pre-activate the endothelium) before tumor cell inoculation (Table VII). In
physiological saline- or VRP + ACV-treated mice (Table VII),
almost all arrested non-Bcl-2-AS-pretreated B16M-F10 cells
appeared as round bright fluorescent cells of well delineated profile
(nondamaged "intact" cells because no fluorescence diffusion from
their cytoplasm to their neighboring tissue was observed).
Bcl-2-AS pretreatment decreased the viability of arrested
metastatic cells (Table VII), which
appeared as irregularly shaped fluorescent cells with a spread of
diffuse fluorescence staining the contiguous hepatic tissue, and were
considered as damaged with cytoplasmic leakage. The decrease in
B16M-F10 viability was sharp in LPS + VRP + ACV-treated mice
inoculated with Bcl-2-AS-pretreated B16M-F10 cells (only 10%
of all arrested cells remained intact) (Table VII).
Nevertheless, it is important to remark that some apparently intact
metastatic cells may have already activated apoptotic mechanisms (see
Fig. 5).
|
|
|
| DISCUSSION |
|---|
B16M-F10 cells surviving the combined nitrosative/oxidative attack
induced by the HSE showed a decrease in GSH content (50).
However, endothelial NO-mediated partial inactivation of -GCS activity is followed by overexpression of -GCS-heavy and -light subunits, which leads to a rapid
recovery of GSH levels within invasive cells (50). Therefore,
as suggested by previous achievements in nonmetastatic models (6), a methodology
capable of maintaining low GSH levels in metastatic cells could
represent a critical advance in cancer therapy.
Here we present evidence showing that GSH is released from highly
metastatic B16M-F10 cells through MRP1 and CFTR (Table I and
Fig. 1).
By using a perifusion system that mimics in vivo conditions,
we show that GSH efflux from B16M-F10 cells can be accelerated
be using Bcl-2-AS (which prevents BCL-2-induced inhibition of
GSH release through CFTR) and VRP (which activates GSH release
through MRP1 (42)) (Tables I
and II).
However, Bcl-2-AS and/or VRP treatment associated with
overexpression of -GCS (Table
IV) increased rates of GSH synthesis
(Table
III) and higher GSH content (Table II) within the B16M-F10 cells.
Recently, we showed that tumor -GT activity, by providing an extra supply of L-cysteine from extracellular GSH, supports GSH
synthesis in B16M-F10 cells and promotes their metastatic growth (4).
Hence, we tested whether inhibition of this activity could prevent
the increase of GSH content within BCL-2-AS- and VRP-treated B16M-F10
cells. Indeed, as shown in Table III, ACV limited GSH synthesis
and allowed GSH efflux to deplete tumor cell GSH. This strategy,
which combines induction of BCL-2 and GSH depletion, sensitized
perifused B16M-F10 cells to the cytotoxic effects of the vascular
endothelium (Tables VI
and VII).
Can possible clinical applications be derived from our study? The
proto-oncogene Bcl-2 and its anti-apoptotic homologs are
mitochondrial membrane permeabilization inhibitors (56)
and participate in the development of chemoresistance (57),
whereas expression of pro-death genes, e.g. Bax or Bak,
is often reduced in cancer cells (58).
In agreement with this idea, Takaoka et al. (59)
observed that Bcl-2 overexpression in B16M cells enhanced
pulmonary metastasis. In fact, a major form of multidrug resistance
in human tumors is caused by overexpression of the MRP1 gene
(7).
In vivo Bcl-2-AS therapy, as explained above, is feasible. In
addition, VRP (at doses that promote a similar plasma concentration
than that used in our experiments) has already been used in
cancer patients with myeloma or acute lymphocytic leukemia, for
example, where it increased accumulation of daunorubicin or
vincristine within the tumor cells (60).
ACV, the L-glutamine analog anti-metabolite, has
followed phase I and II clinical trials in different tumors (61).
Although its use is limited by severe central nervous system
toxicity, a maximum tolerated dose of 50 mg of
acivicin/m2/day has been proposed in combination with the
amino acid solution aminosyn (which decreases drug uptake in the
central nervous system) (61).
In our studies, ACV concentration in the perfusate flow was 1 µM (present only during 30 min) (Fig. 4 and Table III). By taking into account the
circulating blood volume and the in vivo pharmacokinetics in
humans (61),
this means that ACV doses required to block tumor -GT activity will remain within nontoxic levels. This is
important because an increased expression of -GT has been found in melanoma as well as in other cancers
(including human tumors of the liver, lung, breast, and ovary) (62).
In vitro HSE-induced B16M-F10 cytotoxicity was very high (90%)
when Bcl-2-AS-treated metastatic cells were treated with VRP
and ACV (Table VI). These results appear in agreement with
a recent report (63)
showing that GSH depletion enforces the mitochondrial permeability
transition and causes cell death in HL60 cells that overexpress
BCL-2. Furthermore, when BSO- and Bcl-2-AS-pretreated B16M-F10
cells were inoculated intravascularly into mice, the number of intact
arrested cells on the HSE decreased by 98%,
and the very small number of metastatic cell survivors (probably
bearing molecular damages) did not form detectable colonies (5).
Nevertheless, as indicated by the data displayed in Table
VII, after applying in vivo the
methodology proposed in this report, some metastatic cells may still
survive. Invasive cells may prolong survival under dormancy
conditions (64)
or may benefit from oxidative stress-promoting metastatic mechanisms,
e.g. increasing cell adhesion molecule expression (65),
activating early growth response-1 transcription factor gene (66),
activating metalloproteinases (67),
or increasing resistance to oxidative stress (68).
In fact, in mammalian cells at least 40 different gene products are
involved in adaptive responses to oxidative stress (68).
However, invasive B16M-F10 cells that survive after in vitro
interaction with the HSE show a transient impairment of the
mitochondrial system for GSH uptake (50).
The mitochondrial GSH is one of the endogenous effectors that
regulates the mitochondrial permeability transition pore complex (69),
and we observed that B16M-F10 cells with low mitochondrial GSH levels
were highly susceptible to TNF--induced
oxidative stress and death (50).
Most interestingly, this effect can be potentiated by Bcl-2-AS
therapy (5).
Moreover, therapy could be improved by combining nontoxic TNF- doses with IFN- (70),
or with a L-glutamine-enriched diet to
facilitate an L-glutamate-induced inhibition of GSH
transport into tumor mitochondria (71).
Furthermore, our methodology can be combined with cytotoxic drugs
and/or ionizing radiation. Thus the mechanisms described in this
report may have useful applications to improve therapy against
metastatic melanoma and, possibly, against other malignant tumor
types.
| FOOTNOTES |
|---|
.gif) Recipient of a fellowship from the Ministerio de Educación
y Ciencia.
Recipient of a fellowship from the Ministerio de Educación
y Ciencia. ![]()
¶ Recipient of a fellowship from the
Generalitat Valenciana. ![]()
** To whom correspondence should be addressed: Dept. of Physiology, Faculty of Medicine and Odontology, 17 Ave. Blasco Ibañez, 46010 Valencia, Spain. Tel.: 34-963864646; Fax: 34-963864642; E-mail: jose.m.estrela{at}uv.es .
1 The abbreviations used are: B16M,
B16 melanoma; MRP, multidrug resistance protein; Bcl-2-AS,
Bcl-2 antisense oligodeoxynucleotide; -GT, -glutamyl transpeptidase; BSO, L-buthionine-(SR)-sulfoximine; DMEM, Dulbecco's
modified Eagle's medium; PMP, plasma membrane potential; CFTR, cystic
fibrosis transmembrane conductance regulator; G3PDH,
glyceraldehyde-3-phosphate dehydrogenase; VRP, verapamil; ACV,
acivicin; -GCS, -glutamylcysteine synthetase; -GCS-HS, -GCS heavy subunit; -GCS-LS, -GCS light subunit; HSE, hepatic sinusoidal endothelium;
LPS, lipopolysaccharide; PKA, protein kinase A; TUNEL, terminal dUTP
nick-end labeling; BCECF-AM,
2',7'-bis(2-carboxyethyl)-5,6-carboxyfluorescein acetoxymethyl
ester; PBS, phosphate-buffered saline; IFN, interferon; HSE,
hepatic sinusoidal endothelium; TNF, tumor necrosis factor;
ANOVA, analysis of variance. ![]()
| REFERENCES |
|---|
| HOME | HELP | FEEDBACK | SUBSCRIPTIONS | ARCHIVE | SEARCH | TABLE OF CONTENTS |
| All ASBMB Journals | Molecular and Cellular Proteomics |
| Journal of Lipid Research | Biochemistry and Molecular Biology Education |
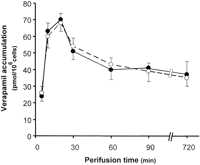
 ) cells was determined over 720 min of perifusion time (see
under "Experimental Procedures"). The concentration of VRP (1 µM in the perfusate flow) was maintained constant
during the perifusion. Data points are means ± S.D. of 4–5
independent determinations.
) cells was determined over 720 min of perifusion time (see
under "Experimental Procedures"). The concentration of VRP (1 µM in the perfusate flow) was maintained constant
during the perifusion. Data points are means ± S.D. of 4–5
independent determinations.