
This site hosts supporting information for PACo: A Novel Procrustes Application to Cophylogenetic Analysis (Balbuena et al. 2013). Although the R scripts provided here are fully functional, if you intend to apply PACo to your data, the recently developed R package paco (Hutchinson et al. 2017) is recommended. A major improvement of the new package over the original scripts is that it provides a large suite of null models by employing the swap algorithms of vegan. In addition, it includes the approach of de Vienne et al. (2011) to handle the transformation of non-Euclidean phylogenetic distance matrices into Principal Coordinates.
Click here for a list of papers citing PACo.
Click here to download paco from R CRAN.
In the Downloads section you will find the R code and examples to implement PACo as described in Balbuena et al. (2013). You will also be able to access the code and tutorial of the Rumbling-Orchids Pipeline to asses divergent evolution between nuclear and organelle sequences as shown in Pérez-Escobar et al. (2016).
|
Contents 1. The Problem 2. Why PACo? 4. Downloads 5. Contact 6. References |
|
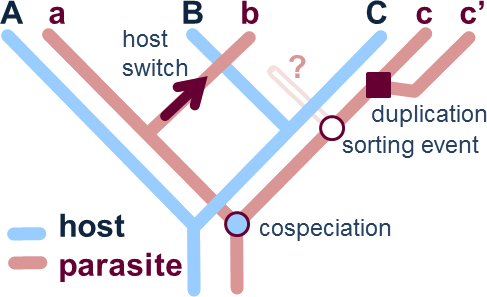
| Figure 1. Example of host and parasite phylogenies illustrating four common evolutionary events contemplated in cophylogenetic studies: cospeciation (hosts and parasites speciate in parallel), host-switch (the parasite is able to colonize a new unrelated host), lineage sorting (failure to speciate or disappearance of a parasite linage on a host lineage) and duplication (independent speciation of the parasite). (Based on Page [1996]). |
1. The Problem
The diversification patterns over evolutionary time of tightly associated organisms, such as parasites and their hosts, are seldom independent. Therefore some degree of congruence (i.e., topological similarity) between the phylogenies of the associated taxa is expected to occur. Congruence expresses the extent to which each node in a given tree maps to a corresponding position in the other tree and perfect congruence can be interpreted as evidence for cospeciation, which may or may not result from coevolutionary mechanisms. Such perfect congruence is rarely, if ever, observed in nature, because in addition to cospeciation, other types of evolutionary events can act concurrently (Fig. 1). Thus, the historical reconstruction of the associations between two given sets of organisms is not straightforward because it needs to evaluate and disentangle the relative roles played by each evolutionary process.
PACo is a global fit method for cophylogenetic analysis based on Procrustes analysis that
- statistically tests and quantifies the congruence the between two given topologies,
- identifies the host-parasite associations contributing most to the cophylogenetic structure.
Although PACo does not explicitly evaluate the contribution of the evolutionary events set forth above, the amount of phylogenetic congruence can be viewed as a measurement of the degree of coevolution in the system studied. For greater usability, PACo can be implemented in the public-domain statistical software R in a reasonable amount of computing time, which affords the analysis of large datasets.
2. Why PACo?
Since there are already many methods for cophylogenetic analysis out there, you might be wondering whether yet another test is really necessary. However, PACo includes several innovative features with respect to previous global-fit methods, such as ParaFit of Legendre et al. (2002) or the cospeciation test described by Hommola et al. (2009):
- PACo is unique in that it produces an informative graphical output for both global evaluation of the fit and assessing the contribution of the individual host-parasite links. In particular, we show in Balbuena et al. (2013) and Pérez-Escobar et al. (2016) that the graphical representation of squared residuals and their confidence intervals is a reasonable alternative to the ParaFitLink1 test (Legendre et al. 2002), enabling more elaborate validations.
- Whereas the previous global fit methods analyse correlation between phylogenies of the associated taxa, PACo is specifically intended for systems where dependence of one phylogeny upon another is assumed. Thus it is ideal to test for the common coevolutionary model that assumes that parasites that spend part of all their life in or on their hosts track the phylogeny of their hosts (Page 1996). Likewise, given that historical area relationships are expected to determine taxa diversification but not the opposite, our method is more appropriate to evaluate diversification of taxa in biogeographical settings. (Nevertheless, the new R package extends this capability and interdependence between two given phylogenies can also be tested if desired.)
- PACo is statistically reliable as shown by its very good performance in terms of Type I and Type II errors. Our simulations indicated superior Type I error performance than ParaFit for the largest phylogenies (20 hosts and 20 parasites) tested. In addition, PACo stands out by its overall higher statistical power.
3. How PACo works
PACo contemplates a given parasite occurring in more than one host species and, conversely, a host harbouring more than one parasite species. Figure 2 gives an overview of the method. The test builds on three pieces of information: two phylogenetic trees corresponding to hosts and parasites, and a binary matrix coding the host-parasite associations (H-P link matrix). Let h and p be the numbers of host and parasite species in the respective phylograms, the H-P link matrix is an h * p matrix, where 1 denotes presence of a given parasite species in a given host species, and 0 corresponds to absence of a particular parasite species in a particular host species.
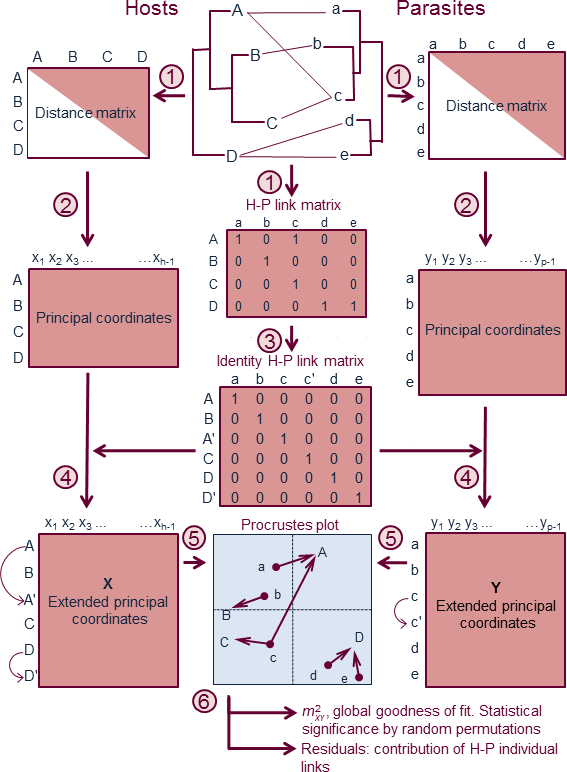 |
| Figure 2. Method overview of PACo: (1) The phylogenetic information encapsulated by the host-parasite (H-P) tanglegram gives way to two distance matrices of host and parasites, and a binary matrix of H-P links. (2) The distance matrices are transformed by Principal Coordinates. (3) The H-P link matrix is converted into an identity matrix to account for multiple host-parasite associations. (4) Rows in the Principal Coordinate matrices are duplicated (arched arrows) following the order dictated by the identity matrix. (5) The extended Principal Coordinate matrices (X and Y) are centred by mean column vectors and subjected to Procrustes analysis, where the parasite configuration is rotated and scaled to fit the host configuration. The fit can be visualised in a Procrustes superimposition plot. (6) The analysis yields a global goodness-of-fit statistic, whose significance can be established by a randomization procedure. The importance of each H-P link can be assessed by the associated squared residual, which together with their 95% confidence intervals, are estimated using a jackknife method. |
4. Downloads
Annotated R code performing PACo, a user guide and example files.
PACo publication and supporting information files (from PLoS ONE).
Example R code of the simulations used in Balbuena et al. (2013). (Useful if you want to develop your own method and wish to compare its performance with PACo, ParaFit and Hommola's et al. [2009]).Rumbling-Orchids Pipeline from Pérez-Escobar et al. (2016).
5. Contact
My fellow coauthors and I will be happy to answer any questions regarding PACo. Suggestions, criticism and feedback will also be most welcome. Please address your queries to j.a.balbuena@uv.es.
6. References
- Balbuena JA, Míguez-Lozano R, Blasco-Costa I (2013) PACo: A Novel Procrustes Application to Cophylogenetic Analysis. PLOS ONE 8(4): e61048).
- de Vienne DM, Aguileta G, Ollier S (2011) Euclidean nature of phylogenetic distance matrices. Syst Biol 60: 826-832.
- Hommola K, Smith JE, Qiu Y, Gilks WR (2009) A permutation test of host-parasite cospeciation. Mol Biol Evol 26: 1457-1468.
- Hutchinson MC, Cagua F, Balbuena JA, Stouffer DB, Poisot T (2017) paco: implementing Procrustean Approach to Cophylogeny in R. Meth Ecol Evol 8: 932-940. doi: 10.1111/2041-210X.12736
- Legendre P, Desdevises Y, Bazin E (2002) A statistical test for host-parasite coevolution. Syst Biol 51: 217-234.
- Page RDM (1996) Temporal congruence revisited: Comparison of mitochondrial DNA sequence divergence in cospeciating pocket gophers and their chewing lice. Syst Biol 45: 151-167.
- Pérez-Escobar OA, Balbuena JA, Gottschling M (2016) Rumbling orchids: How to assess divergent evolution between chloroplast endosymbionts and the nuclear host. Syst Biol 65: 51-65.
Last update: 13 Feb. 2025