Manifestaciones cutáneas de la diabetes
La diabetes mellitus es la enfermedad metabólica más común con un número cada vez mayor de pacientes. Esta enfermedad es un problema de salud mundial y tiene un gran impacto en los sistemas de salud. La diabetes también puede afectar la piel, dando lugar a enfermedades no infecciosas y condiciones y síntomas dermatológicos infecciosos. Cerca de un tercio de todos los pacientes con diabetes tienen algunas manifestaciones cutáneas en el curso de la enfermedad
1. Prurito: El prurito crónico es una manifestación cutánea común de la diabetes. A menudo se asocia con xerosis cutis. Cerca de 3-49% de las personas con diabetes tienen picazón, perjudicando considerablemente su calidad de vida. El prurito crónico puede inducir lesiones de rascado con el desarrollo de lesiones nodulares que se conocen como prurigo nodular. El prurigo nodular consiste en el desarrollo de lesiones nodulares múltiples, localizadas especialmente en las áreas de extensión de extremidades
2. Dermopatía diabética: se caracteriza por el desarrollo de máculas rojizas o parduzcas de <1 cm de diámetro, localizadas especialmente en la cara anterior de las pantorrillas en diabéticos de larga evolución y que parece ser secundaria a la microangiopatía diabética. Afecta tanto a los diabéticos tipo I y II. Las lesiones individuales tienden a presentar remisión espontánea
3. Necrobiosis lipoídica: esta en una dermatosis rara, pero muy característica de la diabetes. Afecta alrededor del 1% de pacientes diabéticos tipo I. Se localiza especialmente en la cara anterior de la pantorrilla, pero puede estar localizada en otras áreas como la cabeza, especialmente en la frente. Consiste en una o múltiples placas rojiza de crecimiento lento, de superficie atrófica, que adquiere una tonalidad amarillenta, rodeada de marcada pigmentación y que con frecuencia se ulcera. Ocasionalmente se observa en pacientes no diabéticos, pero en su mayoría pueden desarrollar la diabetes en el futuro.
4. Granuloma anular: consiste en el desarrollo de placas anulares u ovales , generalmente asintomáticas, especialmente localizadas en el dorso de las articulaciones. El estudio histológico de las lesiones se caracteriza especialmente por un infiltrado intersticial, de predominio histiocitario con formación de granulomas y depósitos de mucina. El granuloma anular puede estar asociado a traumatismos o ser de causa desconocida. Cerca del 20% de casos, especialmente en la forma generalizada está asociada a diabetes.
5. Acantosis nigricans : esta es una alteración frecuente, especialmente en pacientes con obesidad y puede ser indicativa de hiperinsulinemia. Consiste en el desarrollo de piel pigmentada, de superficie verrucosa o aterciopelada, localizada especialmente en los pliegues el cuello, axila y región inguinal. Puede asociar la presencia de acrocordones y fibromas cutáneos. Las causas de la acantosis nigricans pueden ser variadas incluyendo trastornos endocrinos, medicaciones, neoplasia, obesidad o ser un hallazgo aislado. Los pacientes entre 8 y 14 años con acantosis nigricans deben ser valorados para descartar una diabetes o una resistencia a la insulina. La hiperinsulinemia produce una hiperproliferación de los queratinocitos debido a su unión con los factores de crecimiento epidérmico a tipo insulina.
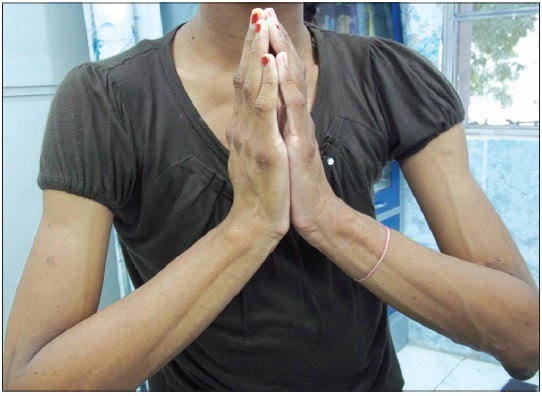
6. Síndrome de la mano diabética o movilidad articular limitada (esclerosis diabética) es una alteración de las manos con rigidez de la palma y de los dedos con resultado de contracturas en flexión y presencia de dedos con piel engrosada y cérea con movilidad reducida. Es una patología frecuente que se observa en el 25-50% de diabéticos, siendo más frecuente en los tipo 1. Generalmente es asintomática, sin embargo, los pacientes con movilidad articular limitada con frecuencia asocian neuropatía. Clínicamente puede ser detectada mediante la realización del signo del orador, solicitando al paciente que disponga sus manos en posición de oración. Un individuo normal es capaz de disponer ambas manos juntas, sin embargo los pacientes con limitación de la movilidad dejan un espacio entre ambas. El cuadro de movilidad articular limitada se relaciona con otras complicaciones de la diabetes incluyendo complicaciones microvasculares (con mayor riesgo de cardiopatía isquémica y enfermedad cerebrovascular), nefropatía, retinopatía, neuropatía,
7. Scleredema diabeticorum de Buschke: El escleredema es una alteración caracterizada por el desarrollo de placas eritematosas, induradas, afectando a la porción superior de la espalda y cuello. Está definido por la presencia de colágeno engrosado y depósitos de mucina intersticial, lo que confiere engrosamiento rigidez y reducción del movimiento de la piel, particularmente del área del hombro. Es una manifestación rara que también puede estar asociada a otras enfermedades sistémicas como paraproteinémias.
8. Bullosis Diabeticorum: Alrededor del 0,5% de pacientes diabéticos desarrollan ampollas en el curso de su enfermedad conocidas como Bullosis diabeticorum. Estos pacientes desarrollan ampollas no inflamatorias de forma espontánea, localizadas en el dorso de extremidades inferiores. La etiología es desconocida y debe realizarse el diagnóstico diferencial con el penfigoide ampolloso.
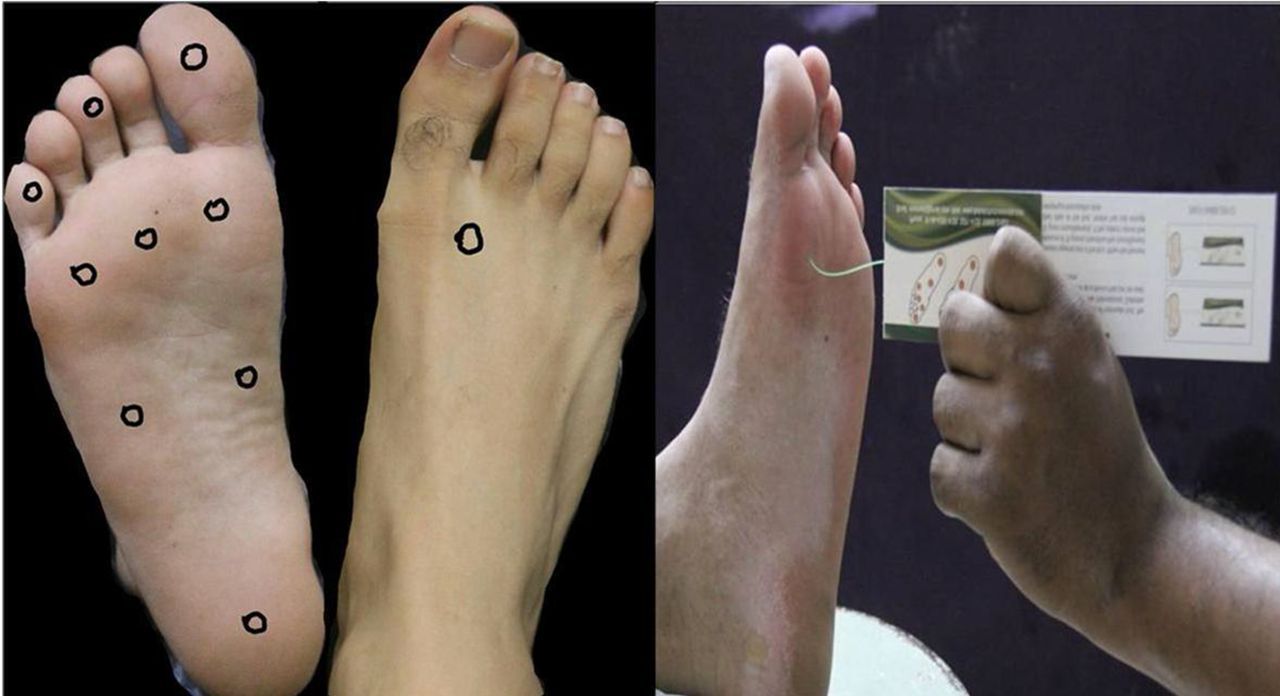
9. Pie diabético: El síndrome del pie diabético afecta a cerca del 6% de diabéticos, compromete seriamente la calidad de vida de los pacientes pues hasta un 1,5% de pacientes con pie diabético va a ser subsidiario de una amputación. La mayoría de las amputaciones pueden ser prevenidas mediante un cuidado adecuado de las complicaciones del pie diabético. La base patogénica del pie diabético es multifactorial. A la aparición del mismo contribuye una diabetes mal controlada que favorece la aparición de neuropatía y enfermedad arterial periférica. La neuropatía especialmente sensitiva facilita la pérdida del estímulo doloroso como factor de protección y facilita la instauración de traumatismos repetidos.
En la prevención del pie diabético es necesario realizar un examen detallado de la presencia de neuropatía diabética y de afectación vascular, así como de la integridad cutánea. El sistema más estandarizado para investigar la neuropatía diabética es la utilización de estudios de sensibilidad mediante la utilización de uno monofilamento de 10 gr. La presencia de neuropatía puede establecerse mediante la palpación de la arteria tibial posterior y la dorsal del pie. Puede realizarse un índice tobillo-braquial que consiste en la relación entre la arteria sistólica entre el tobillo y la medida en el brazo.
Hiperlipidemias
Los xantomas cutáneos son signos de hiperlipidemia. Un xantoma es una colección de histiocitos llenos de lípidos en la piel. Las hiperlipidemias puede ser primarias (genético) o secundarias debido a enfermedades tales como trastornos hepáticos, trastornos pancreáticos, diabetes mellitus, mixedema, síndrome nefrítico, etc. Fármacos, por ejemplo, los retinoides, los glucocorticoides y los estrógenos pueden inducir hiperlipidemia.
Los xantomas varían morfológicamente según el tipo de hiperlipidemia que pueden ser clasificadas en seis grupos. Los xantomas pueden ser localizados, generalizados, en las palmas y las plantas de los pies, sobre los párpados o sobre los tendones.
Tipo de xantomas:
1. Xantelasma es la forma más común de xantoma. Puede estar asociado con un aumento de los lípidos séricos, o pueden encontrarse niveles de lípidos dentro de la normalidad. aparece como placas amarillentas en los párpados.
2. Los xantomas eruptivos son pápulas o placas de color rojo amarillento; suelen aparecer de forma brusca, en todo el cuerpo, pero con mayor frecuencia en las superficies extensoras Se ven en pacientes, con triglicéridos muy elevados.
3. Los xantomas tuberosos son pápulas y nódulos parecidos a los tubérculos, a menudo se encuentra en el codo y las rodillas.
4. Los xantomas tendinosos son nódulos duros y pedregosos en los tendones. se encuentra principalmente en el tendón de Aquiles y los tendones extensores de los dedos. Debido a su profundidad, su color amarillo no se puede apreciar clínicamente Estos a menudo están asociados con hipercolesterolemia grave
5. Los xantomas palmares son xantomas lineales amarillos que aparecen a lo largo de los pliegues palmares.
Porfirias
Las porfirias son trastornos de la síntesis de hemo. Cada tipo de porfiria implica un defecto, heredado o adquirido, en una enzima de la vía. Cuando el defecto es fisiológicamente significativo, se produce una sobreproducción de precursores de la vía que precede al paso defectuoso que pasa a la circulación y se excreta en la orina o la bilis. Las enfermedades se clasifican como porfirias hepáticas (agudas) y porfirias cutáneas. Las porfirias hepáticas se deben a la sobreproducción hepática de los precursores de porfirina, ácido delta aminolevulínico y porfobilinógeno, y los síntomas son causados principalmente por afectación neurológica. La porfiria cutánea se debe a la sobreproducción de porfirinas fotosensibilizadoras por el hígado o la médula ósea. Las porfirias cutáneas más prevalentes son la porfiria cutánea tardía y la protoporfiria. La porfiria aguda intermitente es el tipo agudo más frecuente en la práctica clínica.
Porfiria cutánea tarda
Es la forma más frecuente de porfirias, afecta de 5 a 10 personas por cada 100.000 habitantes. Se debe a la inhibición de la uroporfirinógeno descarboxilasa, la quinta enzima en la ruta biosintética del hemo (Figura 1). El exceso de hierro hepático juega un papel importante en la patogénesis, con más del 50% de los pacientes con porfiria cutánea tarda con una mutación para la hemocromatosis
Patobiología y Manifestaciones: El inicio suele ser después de los 40 años y se caracteriza por la fragilidad de la piel y las lesiones crónicas y ampollas en las áreas expuestas al sol, con mayor frecuencia en la parte posterior de las manos. La fotosensibilidad en la porfiria cutánea tarda se debe a un exceso de porfirinas circulantes, que pasan a un estado de excitación después de la exposición a la luz azul (longitud de onda pico, 410 nm). Con la relajación del estado base anterior, liberan energía que aparece como fluorescencia in vitro y causa daño a la piel. Las mujeres pueden notar el crecimiento del vello facial. El exceso de uroporfirina convierte la orina en marrón o café rojizo. Esencialmente, todos los pacientes con enfermedad clínica tienen al menos dos de los factores de susceptibilidad conocidos, que juntos reducen la actividad de uroporfirinógeno descarboxilasa en aproximadamente 80%. Los pacientes con una mutación de uroporfirinógeno descarboxilasa en un alelo tienen una pérdida de actividad del 50% al inicio del estudio pero tendrá síntomas solo con la inhibición de la enzima residual por uno o más de los factores exógenos (Tabla).
Diagnóstico: Un perfil de porfirina en orina o plasma con predominio de uroporfirina y heptacarboxiporfirina es un diagnóstico de porfiria cutánea tarda, siempre que los niveles de ácido aminolevulínico delta y porfobilinógeno sean normales o solo mínimamente elevados (Tabla). Los pacientes con coproporfiria hereditaria y porfiria variegada pueden presentar síntomas cutáneos similares, pero estas condiciones se pueden distinguir de la porfiria cutánea tardía al medir los niveles de coproporfirina fecal (que son elevados en la coproporfiria hereditaria) y las porfirinas plasmáticas con un pico de emisión de fluorescencia a 626 nm (en porfiria variegata).
La pseudoporfiria es una erupción bullosa que se asemeja a la porfiria cutánea tarda clínicamente pero con niveles normales de porfirinas en plasma y orina. A menudo es idiopática, pero a veces se puede atribuir a medicamentos, especialmente medicamentos antiinflamatorios no esteroideos. No implica exceso de hierro, pero la biopatología es desconocida.
La porfiria eritropoyética congénita de inicio tardío puede simular la porfiria cutánea tarda con respecto a la lesión cutánea y el perfil de porfirinas en la orina. La característica diferenciadora son los niveles marcadamente elevados de porfirinas eritrocíticas en la porfiria eritropoyética congénita.
Tratamiento: La depleción de hierro hepático mediante flebotomía, junto con la restricción de alcohol, tabaco y estrógenos, produce la remisión. Los pacientes que tienen anemia o que tienen efectos adversos con la flebotomía pueden tomar un quelante de hierro oral. El punto final inicial del tratamiento es un nivel de ferritina sérica en el extremo inferior del rango normal (aproximadamente 20 ng por mililitro [454 pmol por litro]), que típicamente se logra con tres a ocho flebotomías. Una alternativa al agotamiento del hierro es una dosis baja de hidroxicloroquina (100 mg) o cloroquina (125 mg), dos veces por semana. Actúan dentro de los hepatocitos para movilizar las porfirinas, que luego se excretan por vía urinaria. Son más convenientes y menos costosas que la flebotomía, y son comparativamente efectivas. Debido a que estos medicamentos no tienen efecto sobre las reservas de hierro hepático, se prefiere la flebotomía para los pacientes con hemocromatosis genética. Dos tercios de los pacientes con porfiria cutánea tardía tienen infección por el virus de la hepatitis C (VHC). Los informes de casos han sugerido que la erradicación del virus conduce a la resolución de la enfermedad de la piel.
Pronóstico: La enfermedad responde al tratamiento inicial en al menos el 90% de los casos, pero puede recurrir. Los pacientes con hemocromatosis genética requieren una flebotomía periódica para mantener el nivel de ferritina sérica por debajo de 100 ng por mililitro (225 pmol por litro). Los pacientes que consumen más de cuatro bebidas alcohólicas al día o continúan fumando pueden recaer. Sin embargo, las mujeres que requieren terapia de reemplazo hormonal pueden continuar recibiendo estrógenos transdérmicos después de la remisión. En todos los pacientes, se recomienda una revisión anual de los niveles de uroporfirina en orina o plasma para la detección temprana de la recidiva y para el retratamiento.
PORFIRIA AGUDA INTERMITENTE
La porfiria intermitente aguda se debe a la deficiencia parcial de la tercera enzima de la síntesis de hemo, porfobilinógeno desaminasa. Es de herencia autosómica dominante. La prevalencia de mutaciones en las poblaciones occidentales es de aproximadamente 1 portador por cada 2000 personas. Sin embargo, los ataques agudos ocurren en menos del 10% de la población en riesgo; esto refleja un papel clave de los factores ambientales y posiblemente modificadores genéticos.
Manifestaciones: El paciente típico con un ataque de porfiria intermitente aguda es una mujer joven previamente sana que ha tenido varios días de fatiga severa e incapacidad para concentrarse, seguido de un empeoramiento progresivo del dolor abdominal, náuseas, vómitos y signos neurológicos sutiles (debilidad, disestesia y afecto alterado). Los agentes analgésicos, incluidos los opioides, han proporcionado poco o ningún alivio. La historia clínica del paciente puede revelar visitas previas al servicio de urgencias con los mismos síntomas y una evaluación no diagnóstica. Se acompaña de afectación sistémica con taquicardia y la presión arterial sistólica elevada. Las imágenes abdominales pueden mostrar cambios consistentes con el íleo pero por lo demás son normales. La falta de hallazgos objetivos y una respuesta deficiente a los analgésicos a menudo crean una impresión inicial de dolor psicosomático o adicción a las drogas. La tríada de convulsiones, dolor abdominal e hiponatremia en una mujer joven es altamente sugestiva de porfiria aguda.
Diagnóstico: Los niveles elevados de porfobilinógeno en orina o plasma son específicos para la porfiria aguda., alcanzando de 10 a 150 veces el límite superior del rango normal .Hoy en día, el único dato para un diagnóstico confirmado es una prueba de porfobilinógeno
Tratamiento: El tratamiento inicial incluye la revisión de los medicamentos del paciente para cualquiera que se considere de riesgo en pacientes con porfiria. Actualmente, el único tratamiento específico para los ataques agudos es el hemo intravenoso (Panhematin en los Estados Unidos y Normosang en Europa).
El manejo de la porfiria intermitente aguda también involucra la detección genética de los miembros de la familia, especialmente los padres, hermanos e hijos del paciente. Se aconseja a los miembros de la familia que son portadores de la mutación que eviten un ataque agudo y la importancia de evaluar a la próxima generación. Se les puede asegurar que la mayoría del riesgo de un ataque agudo está asociado con factores ambientales manejables. En más de 400 mutaciones identificadas, hay poca evidencia de que el genotipo prediga el fenotipo.8
Pronóstico: Antes de la década de 1980, la mortalidad entre los pacientes con ataques agudos de porfiria era aproximadamente del 25%. Con el diagnóstico precoz y el tratamiento específico, la perspectiva ha mejorado mucho. Cuando la enfermedad de la neurona motora es parte de un ataque agudo, la debilidad asociada se resuelve lentamente, pero generalmente por completo durante un período de 1 año; ocasionalmente, la caída del pie o la caída de la muñeca no se resuelven durante el período de recuperación. Los estudios con pacientes mayores con porfiria intermitente aguda han mostrado una mayor prevalencia de enfermedad hepática crónica y enfermedad renal, posiblemente debido a los efectos tóxicos a largo plazo del ácido aminolevulínico delta.El riesgo de daño renal puede estar relacionado con la variación genética en el péptido transportador 2, que transporta ácido delta aminolevulínico así como péptidos cortos. Aunque la enfermedad hepática rara vez alcanza el estadio de la cirrosis, un estudio sueco indicó que el riesgo de cáncer primario de hígado (principalmente carcinoma hepatocelular) aumenta en un factor de 80 después de la 50 años de edad (y por un factor de 150 en mujeres); Se recomienda el cribado anual con medición del nivel de alfa-fetoproteína y la obtención de imágenes abdominales (por ejemplo, ecografía) 39.administración del vector.
Protoporfiria
La protoporfiria se debe a la sobreproducción de protoporfirina por la médula ósea. Hay dos formas, cada una de las cuales es causada por una mutación genética distinta: deficiencia de ferroquelatasa, que da lugar a la protoporfiria eritropoyética "clásica"; y la hiperactividad delta del ácido aminolevulínico sintasa 2, denominada protoporfiria ligada a X (debido a que el gen que codifica la ácido delta aminolevulínico sintasa 2 se localiza en el cromosoma X). La protoporfirina circulante se excreta completamente en la bilis; por lo tanto, el color de la orina es normal.
Manifestaciones: Los síntomas de la protoporfiria, que generalmente comienzan en la primera infancia cuando los niños pequeños se exponen al aire libre, consisten en picor intenso, ardor y picazón en la piel expuesta al sol. El inicio después de la exposición al sol varía, pero puede ser de 10 minutos. Los niños mayores y adultos, particularmente aquellos con piel más oscura, pueden manejar estar afuera por una hora o más. Debido a que la piel de un bebé muestra poco más que inflamación leve y eritema, el problema puede denominarse "alergia al sol", y la protoporfiria puede no diagnosticarse hasta años después. Con lesiones repetidas, se puede desarrollar hiperqueratosis leve y liquenificación sobre los nudillos y alrededor de la boca.
Herencia: En la mayoría de los pacientes, solo uno de los dos alelos de la ferroquelatasa está mutado. Debido a que la expresión clínica varía ampliamente, la enfermedad se clasificó como autosómica dominante con penetrancia variable. Sin embargo, la pérdida de actividad enzimática en casos sintomáticos excede el 50%, lo que no es consistente con una mutación de un solo alelo. En algunas familias, el patrón de herencia es autosómico recesivo.
{kind=link}
.jpg){kind=link}
{kind=link}