|
|
|
|
A. Soriano, E. Silla, I. Tuñón The dissociative electron transfer reaction CH3Cl + e- > CH3· + Cl- in aqueous solution is studied by using a QM/MM method. In this work the quantum subsystem (a methylchloride molecule plus an electron) is described using Density Functional theory while the solvent (300 water molecules) is described using the TIP3P classical potential. By means of Molecular Dynamics simulations and the thermodynamic integration technique we obtained the Potential of Mean Force (PMF) for the carbon-chlorine bond dissociation of the neutral and radical anion species. Combining these two free energy curves we found a quadratic dependence of the activation free energy on the reaction free energy in agreement with Marcus’ relationship, originally developed for electron transfer processes not involving bond breaking. We also investigated dynamical aspects by means of 60 dissociative trajectories started with the addition of an extra electron to different configurations of a methylchloride molecule in solution. The PMF shows the existence of a very flat region, in which the system is trapped during some finite time if the quantum subsystem quickly losses its excess kinetic energy transferring it to the solvent molecules. One of the most important factors determining the effectiveness of this energy transfer seems to be the existence of close contacts (hydrogen bonds) between the solute and the solvent.
R. Castillo, E. Silla, I. Tuñón We present a theoretical study of a mechanism for the hydrolysis of the acyl-enzyme complex formed by a class A ß-lactamase (TEM1) and an antibiotic (penicillanate), as a part of the process of antibiotic’s inactivation by this type of enzymes. In the presented mechanism the carboxylate group of a particular residue (Glu166) activates a water molecule accepting one of its protons and afterwards transfer this proton directly to the acylated serine residue (Ser70). In our study we employed a quantum mechanics (AM1)-molecular mechanics partition scheme (QM/MM) where all the atoms of the system were allowed to relax. For this purpose we used the GRACE procedure in which part of the system is used to define the Hessian matrix while the rest is relaxed at each step of the stationary structures search. Using this computational scheme, the hydrolysis of the acyl-enzyme is described as a three-steps process: The first one corresponds to the proton transfer from the hydrolytic water molecule to the carboxylate group of Glu166 and the subsequent formation of a tetrahedral adduct as a consequence of the attack of this activated water molecule to the carbonyl carbon atom of the ß-lactam. In the second one, the acyl-enzyme bond is broken obtaining a negatively charged Ser70. In the last step this residue is protonated by means of a direct proton transfer from Glu166. The rate-determining step at our computational level is the acyl-enzyme bond breaking leading to the second reaction intermediate. The large mobility of Glu166, a residue that is placed in a W-loop, is essential to facilitate this mechanism. The geometry of the acyl-enzyme complex shows a large distance between Glu166 and Ser70 and thus, if protein coordinates were kept frozen during the reaction path, it would be difficult to get a direct proton transfer between these two residues .
I. Tuñón, E. Silla, M. F. Ruiz-López The experimental activation energy for the tautomerization of glycine (zwitterion ® neutral form) has been reported to be 14.6 kcal/mol. It has been generally assumed that this energy barrier is needed for proton transfer to occur. However, previous theoretical results do not support this interpretation. In the present work, we examine this question using density functional calculations, extended basis sets and a polarizable continuum solvent model. Our results suggest that the limiting step for the tautomerization process corresponds basically to H-atom reorientation in the -COOH group. This could be a general feature in the tautomerization of aminoacids.
F. R. Tortonda, E. Silla, I. Tuñón, D. Rinaldi, M. F.
Ruiz-López Serine aminoacid in aqueous solution is theoretically studied at the B3PW91/6-31+G** level using a dielectric continuum solvent model. Neutral and zwitterionic structures in gas phase and solution are described and the proton transfer mechanism is discussed. A neutral conformation in which the carboxyl hydrogen atom is already oriented towards the amino group seems to be the absolute energy minimum in the gas phase and the most stable neutral form in solution. The absolute energy minimum in solution is a zwitterionic form. The energy barrier for proton transfer is predicted to be very small, in particular when zero-point contributions are added. Our calculations allow to discuss the dynamic aspects of the ionisation mechanism by incorporating non-equilibrium effects.
S. Martí, J. Andrés, V. Moliner, E. Silla, I. Tuñón, J. Bertrán. Preorganization and Reorganization as Related Factors in Enzyme Catalysis. The Chorismate Mutase Case. Chem. Eur. J. 9, 984-991 (2003). In this paper a deeper insight into the chorismate to prephenate rearrangement, catalyzed by the Bacillus subtilis Chorismate Mutase, is provided by means of a combination of statistical Quantum Mechanics/Molecular Mechanics simulation methods and hybrid Potential Energy Surface exploration techniques. The main aim of this work is to present an estimation of the preorganization and reorganization terms of the enzyme catalytic rate enhancement. For analyzing the first one we have studied different conformational equilibria of chorismate in aqueous solution and in the enzyme active site. Our conclusion is that Chorismate Mutase preferentially binds the reactive conformer of the substrate, the one presenting a structure similar to the transition state of the reaction to be catalyzed, with shorter distances between the carbon atoms to be bonded and more diaxial character. With respect to the reorganization effect, an energy decomposition analysis of the potential energy of the reactive reactant and the reaction transition state in aqueous solution and in the enzyme shows that the enzyme structure is better adapted to the transition structure. This means not only a more negative electrostatic interaction energy with the transition state but also a low enzyme deformation contribution to the energy barrier. Our calculations reveal that the structure of the enzyme is responsible of stabilizing the transition state structure of the reaction, with concomitant selection of the reactive form of the reactants. This is, the same enzymatic pattern that stabilizes the transition structure promotes those reactant structures closer to the transition structure; i.e. the reactive reactants. In fact, both reorganization and preorganization effects have to be considered as the two faces of the same coin, having a common origin in the effect of the enzyme structure on the energy surface of the substrate. Snapshot of Transition Structure in the BsCM active site
F. J. Ramírez, I. Tuñón, J. A. Collado, E. Silla. Structural and Vibrational Study of the Tautomerism of Histamine Free-Base in Solution. J. Am. Chem. Soc. 125, 2328-2340 (2003). Infrared and Raman spectroscopy in H2O and D2O and quantum Density Functional calculations were used to determine the structure of histamine free-base in aqueous solution. A quantum mechanical study of the tautomeric equilibrium of histamine free-base in solution were performed at the 6-311G** level. Electronic correlation energies were included by using the hybrid functional B3LYP. The solvent was simulated as a continuum characterised by a dielectric constant, and the quantum system (solute) was placed in an ellipsoidal cavity. Solute-solvent electrostatic interaction was calculated by means a multipolar moment expansion introduced in the Hamiltonian. Four relevant histamine conformations were optimised by allowing all the geometrical parameters to vary independently, which involved both the gauche-trans and the N3H-N1H tautomerisms. The calculated free energies predicted N3H-gauche as the most stable one of histamine free-base in solution. The order of stability is here completely altered respect to previous results in gas phase, which presented the N1H-gauche conformer as the most stable structure. Our results also differ from previous Monte Carlo simulations, which obtained the N3H-trans conformer as the most stable in solution, although in this case the histamine structures were kept frozen to the gas phase geometry. Vibrational spectroscopy results support theoretical ones. Quadratic force fields for the four histamine conformers were achieved under the same calculation methodology. Previously, a general assignment of the infrared and Raman spectra of histamine free-base was proposed for solutions in both natural and heavy water. This allowed us to compare the experimental set of both wavenumbers and infrared intensities with the calculated ones. The lowest quadratic mean wavenumber deviation was obtained for the N3H-gauche conformer, in agreement with the free-energy calculations. Calculated infrared intensities were also compared to the experimental intensities, supporting this conformer as the relevant structure of histamine free-base in solution. It was then selected for a complete vibrational dynamics calculation, starting by a low-level scaling procedure to fit the set of calculated wavenumbers to the experimental values. The results were presented in terms of quadratic force constants, potential energy distribution and normal modes. Infrared intensities were also computed and successfully compared to the observed intensities.
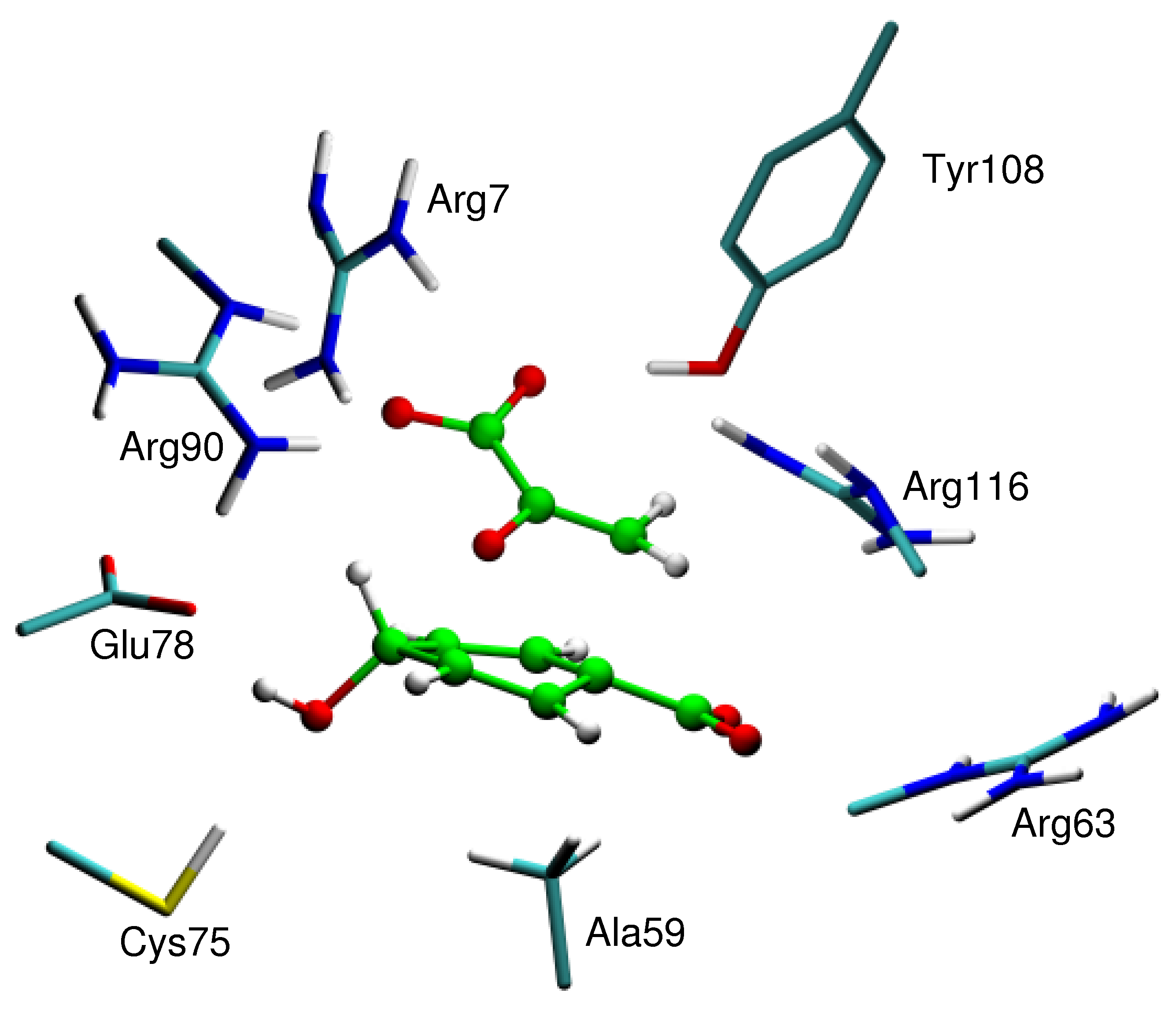
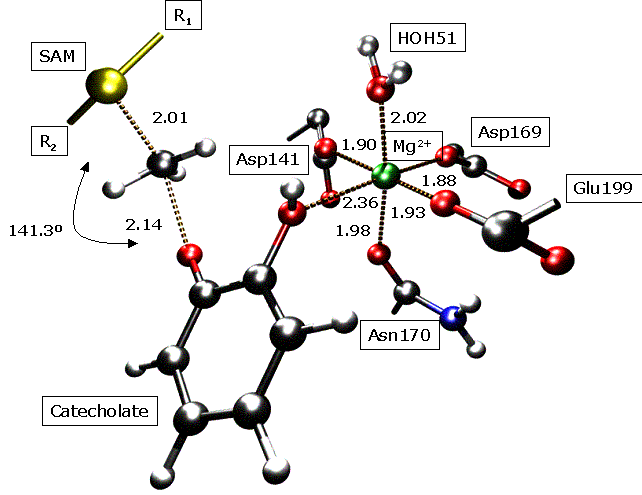
M. Roca, S. Martí, J. Andrés, V. Moliner, I. Tuñón, J. Bertrán, I. H. Williams. Theoretical Modelling of Enzyme Catalytic Power: Analysis of Cratic and Electrostatic Factors in Cathecol O-MethylTransferase. J. Am. Chem. Soc. 125, 7726-7737 (2003). A comparative theoretical study of a bimolecular reaction in aqueous solution and catalyzed by the enzyme catechol O-methyl transferase (COMT) has been carried out by a combination of two hybrid QM/MM techniques: statistical simulation methods and internal energy minimizations. In contrast to previous studies by other workers, we have located and characterized transition structures for the reaction in the enzyme active site, in water and in vacuum, and our potential of mean force calculations are based upon reaction coordinates obtained from features of the potential energy surfaces in the condensed media, not from the gas phase. The AM1/CHARMM calculated free energy of activation for the reaction of S-adenosyl methionine (SAM) with catecholate catalyzed by COMT is 15 kcal mol-1 lower the AM1/TIP3P free energy barrier for the reaction of trimethylsulfonium cation with catecholate anion in water at 300 K, in agreement with previous estimates. The thermodynamically preferred form of the reactants in the uncatalyzed model reaction in water is a solvent-separated ion-pair (SSIP). Conversion of the SSIP into a contact ion-pair, with a structure resembling that of the Michaelis complex (MC) for the reaction in the COMT active site, is unfavourable by 7 kcal mol-1, largely due to reorganization of the solvent. We have considered alternative ways to estimate the so-called “cratic” free energy for bringing the reactant species together in the correct orientation for reaction, but conclude that direct evaluation of the free energy of association by means of molecular dynamics simulation with a simple standard-state correction is probably the best approach. The latter correction allows for the fact that the size of the unit cell employed with the periodic boundary simulations does not correspond to the standard state concentration of 1 M. Consideration of MC-like species allows a helpful decomposition of the catalytic effect into preorganization and reorganization phases. In the preorganization phase the substrates are brought together into the MC-like species, either in water or in the enzyme active site. In the reorganization phase the roles of the enzymic and aqueous environments may be compared directly because reorganization of the substrate is about the same in both cases. Analysis of the electric field along the reaction coordinate demonstrates that in water the TS is destabilized with respect to the MC-like species because the polarity of the solute diminishes and consequently the reaction field is also decreased. In the enzyme the electric field is mainly a permanent field and consequently there is only a small reorganization of the environment. Therefore destabilisation of the TS is lower than in solution and the activation barrier is smaller.
|