.svg)




A key mechanism in the evolution of human tuberculosis is being investigated
- Scientific Culture and Innovation Unit
- September 20th, 2019
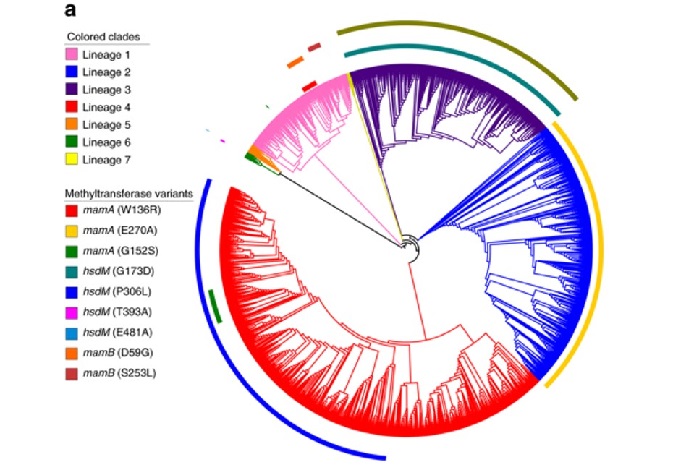
MTBC phylogeny with 4595 genomes from all over the world including the seven known human lineages. Each arc represents a mutation that has inactivated a methylation pattern in the corresponding strains. /CSIC.
Research staff from the Institute for Integrative Systems Biology (I2SysBio), a joint centre of the CSIC and the Universitat de València; the Biomechanics Institute of València (IBV) and the Fundación para el Fomento de la Investigación Sanitaria y Biomédica (FISABIO, Foundation for the Promotion of Health and Biomedical Research), among others, have studied the effect that mutations have on the different strains of tuberculosis. The results, published in the journal Nature Communications, broaden the knowledge about the evolution of the bacteria causing this disease in humans.
Tuberculosis is an infectious disease caused by the Mycobacterium tuberculosis bacteria, that causes a devastating mortality in humans and animals, and also entails significant economic losses. Knowing how different bacterial lineages differ increases understanding of the origins of the bacteria causing the disease and the genetic mechanisms involved.
Álvaro Chiner Oms, researcher at the Mixed Infection and Public Health Unit (Institute for Integrative Systems Biology, FISABIO-CSISP) and the IBV, points out that “we have carried out an exhaustive comparison of transcriptomes and DNA-methyl's of nineteen isolated strains representative of the global phylogenetic spectrum of MTBC strains adapted to humans, which has allowed us to discover unique transcriptional profiles”.
Iñaki Comas, also a CSIC researcher at IBV, explains that in non-recombinant bacteria, where the mutation provides most of the genetic variation, “selective and non-selective processes can have a great impact on functional diversification. Some of these functional differences can be translated into phenotypic characteristics. The complex Mycobacterium tuberculosis or MTBC, which comprises a group of mycobacteria that affect humans and some mammalian species, despite their extremely low diversity, shows important biological differences between strains and phylogenetic lineages”.
In addition, the expert states: “we have observed that the patterns of differential transcription between lineages reflected the constitutive expression of genes that are normally associated with the virulence of the bacterium. Isolated from the opportunity to generate diversity through horizontal gene transfer, transcriptional adaptation may allow isolated strains of MTBC to optimize their infectivity and transmission in subtly different environments provided by different human host populations.” In this sense, Comas concludes: “our study demonstrates that variation in MTBC transcriptional profiles is primarily due to mutations at the point of transcription initiation and have probably evolved due to differences in host characteristics.”
This discovery could improve knowledge of how different strains of tuberculosis have adapted to human populations to be successfully transmitted to the present day.
Article:
Álvaro Chiner-Oms, Michael Berney, Christine Boinett, Fernando González-Candelas, Douglas Young, Sebastien Gagneux, William R. Jacobs Jr., Julian Parkhill, Teresa Cortes i Iñaki Comas. «Genome-wide mutational biases fuel transcriptional diversity in the Mycobacterium tuberculosis complex». Nature Communications. DOI: https://doi.org/10.1038/s41467-019-11948-6
File in: Recerca, innovació i transferència , Institut de Biologia Integrativa de Sistemes (I2SYSBIO) , Facultat de Ciències Biològiques , Internacionalització recerca , Investigació a la UV , Grups de recerca
Other News



Tags
Covid
Culture
Studies
Institutional
Organisation
People
Research, innovation and transfer
University and Society
Living at the University