![]()
Análisis de agrupamiento iterativo de datos de interacción proteíca
English version
| |
|
Análisis de agrupamiento iterativo de datos de interacción proteíca |
English version |
||||
|
|
|||||
|
|
|||||
|
![]() Utilizando UVCLUSTER
Utilizando UVCLUSTER
UVCLUSTER debe ejecutarse en una ventana de comandos, usando la siguiente sintaxis: uvcluster NN AC Donde: NN = Número de soluciones aleatorias1 AC = Coeficiente de afinidad2 [1-100]
1.- Es recomendable utilizar el menos 10 veces el número de elementos. 2.- Afecta al rigor del análisis, ver Arnau et al. 2004 para más detalles.
|
![]()
![]() Diagrama de flujo del UVCLUSTER
Diagrama de flujo del UVCLUSTER
 |
|
|
|
< |
Los análisis del UVCLUSTER se inician importando un fichero de texto que contenga una relación de interacciones directas proteína-proteína. | |
|
< |
Primer filtro optativo: Usar sólo las interacciones entre pares de proteínas de una lista o excluir todas aquellas interacciones que contengan alguna proteína de la lista. | |
|
< |
Segundo filtro optativo: Establecer un límite para el máximo/mínimo número de interacciones requeridas para incluir una proteína en el análisis. | |
|
< |
Generación y grabación de la matriz de distancias primarias (que puede
usarse subsiguientemente como entrada para el UVCLUSTER). Dos archivos, con extensiones: .pro y .tab |
|
|
< |
Selección de las proteínas de interés: Mediante una lista o escogiendo todas la proteínas a distancia N de una dada. | |
|
< |
Generación de la matriz de distancias secundarias y grabación de los ficheros de salida. |
![]()
![]() Output files
Output files
|
Los ficheros se nombran automáticamente de acuerdo con la siguiente fórmula: (Selección)_(Nombre de la matriz de distancias primarias empleada)_NN_AC# Añadiendo respectivamente: "_S1.txt", "_S2.txt", ".pgm" or "_pgmpro.txt" (Selección) puede ser: (Proteína escogida)_(Distancia)_ (Nombre de la lista de proteínas)_
|
|
Fichero de salida S1: |
El primer fichero de salida contiene las tablas de distancias primarias y secundarias entre los elementos escogidos, además de los valores de parámetros significativos empleados en el análisis. También incluye una tabla de distancias secundarias con el formato adecuado para ser copiada en un fichero de texto e importada a MEGA 2.1. |
| Fichero de salida S2: |
El segundo fichero de salida muestra los resultados de un agrupamiento jerárquico aglomerativo usando UPGMA, empleando los datos de distancias secundarias. |
| Ficheros de salida PGM: |
El tercer fichero de salida es una representación gráfica de los datos en PGM (Portable Grey Map), consistente en un cuadrado formado por K2 cuadrados menores cuya intensidad indica el grado de interacción entre cada par de proteínas. Los archivos PGMA pueden abrirse con programas freeware como IrfanView. El orden de las proteínas en la matriz está optimizado para resaltar los clústers. Esta ordenación se proporciona en el fichero *_pgmpro.txt y corresponde a un agrupamiento UPGMA. |
![]()
![]() Velocidad de ejecución
Velocidad de ejecución
|
Todos los tiempos han sido obtenidos en un PC estándard (procesador Intel Pentium IV a 2.8 GHz, con 515 MB de RAM). |
|
Todos los datos disponibles en enero de 2004 en la base de datos DIP sobre S. cerevisiae (4721 proteínas, 15210 interacciones), fueron transformados en una tabla de distancias primarias en 14 minutos. Esta matriz de distancias fue la utilizada para proporcionar los datos de entrada para los análisis. El tiempo se incrementa linealmente con el número de iteraciones, también se incrementa, pero de forma no lineal, al disminuir los valores de AC. |
|
Elementos considerados |
Nº iteraciones |
AC = 100 |
AC = 50 |
|
34 elementos y 561 distancias primarias (ver ejemplo para más detalles) |
10,000 | < 2 seg. | < 2 seg. |
|
34 elementos y 561 distancias primarias (ver ejemplo para más detalles) |
100,000 | 9 seg. | 13 seg. |
|
150 proteínas escogidas al azar (11.175 distancias primarias) |
10,000 | 9 seg. | 125 seg. |
|
500 proteínas escogidas al azar (124.750 distancias primarias) |
10,000 | 23 min. | 160 min. |
![]()
|
|
|
El grafo siguiente ejemplifica el problema de los empates en proximidad, inherente a las redes de interacción proteíca, que puede ser abordado utilizando la estrategia de agrupamiento jerárquico del UVCLUSTER. Dos clústers (unidades 1-4 y 8-11) resultan patentes. |
|
|
|
A continuación, se presenta un árbol UPGMA obtenido empleando distancias primarias que claramente fracasa a la hora de detectar los dos clústers. Este error acontecerá siembre que los empates se resuelvan de forma tal que, por azar, las unidades 4 y 5 (o, alternativamente, 7-8) resulten agrupadas. |
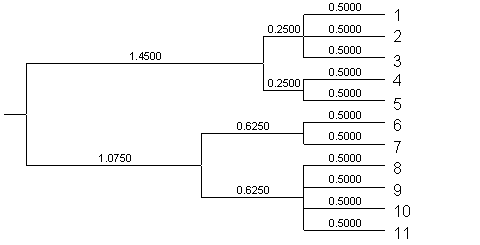 |
|
Al aplicar el algoritmo UPGMA a las distancias secundarias obtenidas mediante UVCLUSTER, con N=10000 y AC=100, la topología del árbol se corresponde claramente con el grafo. Las distancias entre las unidades 1-3 o 9-11 son iguales a cero, y las unidades 4 y 8 son, del resto, las conectadas a menor distancia. |
 |
 |
Fichero de salida PGM.. El orden e las proteínas en la matriz es el siguiente: 1, 2, 3, 4, 11, 10, 9, 8, 6, 5 y 7. Cuento más claro más próximas. |
![]()
