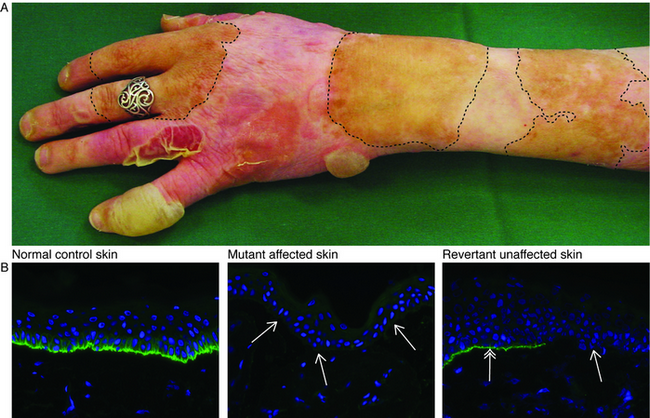
igure 1. A. A patient with non-Herlitz junctional epidermolysis bullosa caused by recessive mutations in the COL17A1 gene: a frameshift mutation in exon 18 (c.1601delA) and a nonsense mutation in exon 51 (c.3676C>T). Due to absence of type XVII collagen (Col17), her skin is hypopigmented and blisters easily. She has, however, several areas of skin that are clinically normal and do not blister. The (hyper)pigmented areas are indicated by the dashed line. B. Skin biopsies taken from the affected skin show the absence of Col17 staining (green) in the basement membrane zone (arrows) compared to normal control skin. In contrast, skin biopsies taken from the normal appearing skin show patchy re-expression of the deficient protein. In this biopsy of revertant skin a mosaic pattern was observed with the left side a stretch containing revertant cells (double arrowhead) and the right side uncorrected basal cells (single arrowhead; patient described in Jonkman et al., 1997; Pasmooij et al., 2005).
Peter C. van den Akker, MD; Miranda Nijenhuis, BAS; Gonnie Meijer; Robert M. W. Hofstra, PhD; Marcel F. Jonkman, MD, PhD; Anna M. G. Pasmooij, PhD
Dystrophic epidermolysis bullosa is a genetic blistering disorder caused by mutations in the type VII collagen gene, COL7A1. In revertant mosaicism, germline mutations are corrected by somatic events resulting in a mosaic disease distribution. This “natural gene therapy ” phenomenon long has been recognized in other forms of epidermolysis bullosa but only recently in dystrophic epidermolysis bullosa.
We describe a 21-year-old man with recessive dystrophic epidermolysis bullosa carrying the homozygous c.6508C>T (p.Gln2170X) nonsense mutation who reported an unaffected skin patch on his neck where blisters never had occurred. Immunofluorescent type VII collagen staining was normal in 80% of the unaffected skin biopsy; however, it was strongly reduced in the affected skin. In the unaffected skin, the somatic nucleotide substitution c.6510G>T reverted the germline nonsense codon to tyrosine (p.Gln2170Tyr), thereby restoring functional protein production.
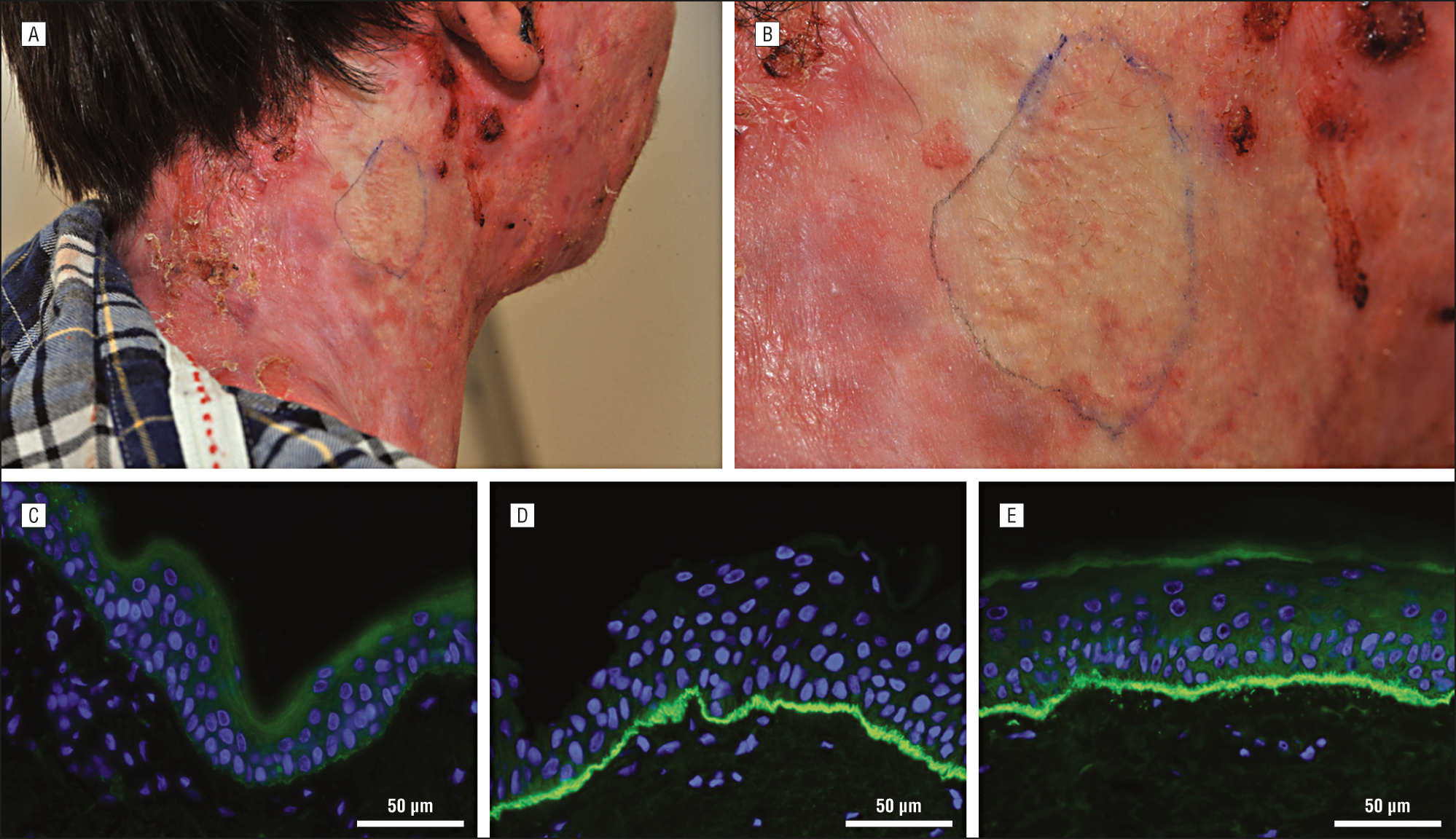
Revertant mosaicism is considered rare in recessive dystrophic epidermolysis bullosa. However, it might be more common than previously anticipated because our patient is the third in whom revertant mosaicism was identified in a short period of time. The correction mechanism is different than that previously reported. Systematic examination of patients with recessive dystrophic epidermolysis bullosa, therefore, will likely reveal more patients with revertant patches. This is important because the natural gene therapy phenomenon may provide opportunities for revertant cell therapy.