Publications
Featured
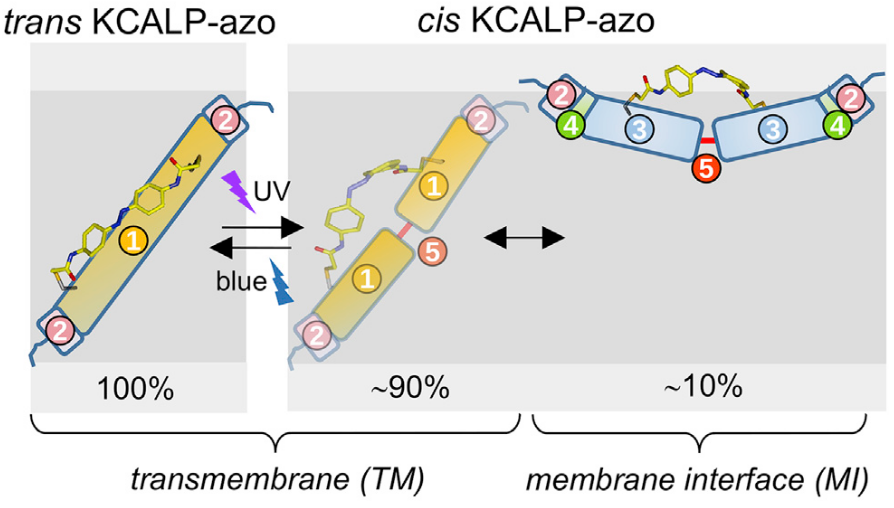
A photoswitchable helical peptide with light-controllable interface/transmembrane topology in lipidic membranes
Gutiérrez-Salazar M, Santamaría-Aranda E, Schaar L, Salgado J, Sampedro D & Lorenz-Fonfria VA
iScience (2021)
24: 102771.
Tweet
Highlights: We present an α-helical transmembrane peptide modified with a molecular photoswitch. The peptide exhibits reversible photocontrol of its membrane topology. A fraction moves to the membrane interface with UV and inserts back with blue light. This system will be useful to address the molecular mechanism for membrane insertion.
The spontaneous insertion of helical transmembrane (TM) polypeptides into lipid bilayers is driven by three sequential equilibria: solution-to-membrane interface (MI) partition, unstructured-to-helical folding, and MI-to-TM helix insertion. A bottleneck for understanding these three steps is the lack of experimental approaches to perturb membrane-bound hydrophobic polypeptides out of equilibrium rapidly and reversibly. Here, we report on a 24-residues-long hydrophobic α-helical polypeptide, covalently coupled to an azobenzene photoswitch (KCALP-azo), which displays a light-controllable TM/MI equilibrium in hydrated lipid bilayers. FTIR spectroscopy reveals that trans KCALP-azo folds as a TM α-helix (TM topology). After trans-to-cis photoisomerization of the azobenzene moiety with UV light (reversed with blue light), the helical structure of KCALP-azo is maintained, but its helix tilt increased from 32 ± 5° to 79 ± 8°, indication of a reversible TM-to-MI transition. Further analysis indicates that this transition is incomplete, with cis KCALP-azo existing in a 90% TM and 10% MI mixture.
All publications
2023-24
- Experimental and computational biophysics to identify vasodilator drugs targeted at TRPV2 using agonists based on the probenecid scaffold., Catalina-Hernández È, López-Martín M, Masnou-Sánchez D, Martins M, Lorenz-Fonfria VA, Jiménez-Altayó F, Hellmich UA, Inada H, Alcaraz A, Furutani Y, Nonell-Canals A, Vázquez-Ibar JL, Domene C, Gaudet R & Perálvarez-Marín A.
Comput Struct Biotechnol J (2024)
23: 473-482
.
TRP channels are important pharmacological targets in physiopathology. TRPV2 plays distinct roles in cardiac and neuromuscular function, immunity, and metabolism, and is associated with pathologies like muscular dystrophy and cancer. However, TRPV2 pharmacology is unspecific and scarce at best. Using in silico similarity-based chemoinformatics we obtained a set of 270 potential hits for TRPV2 categorized into families based on chemical nature and similarity. Docking the compounds on available rat TRPV2 structures allowed the clustering of drug families in specific ligand binding sites. Starting from a probenecid docking pose in the piperlongumine binding site and using a Gaussian accelerated molecular dynamics approach we have assigned a putative probenecid binding site. In parallel, we measured the EC50 of 7 probenecid derivatives on TRPV2 expressed in using a novel medium-throughput Ca influx assay in yeast membranes together with an unbiased and unsupervised data analysis method. We found that 4-(piperidine-1-sulfonyl)-benzoic acid had a better EC50 than probenecid, which is one of the most specific TRPV2 agonists to date. Exploring the TRPV2-dependent anti-hypertensive potential in vivo, we found that 4-(piperidine-1-sulfonyl)-benzoic acid shows a sex-biased vasodilator effect producing larger vascular relaxations in female mice. Overall, this study expands the pharmacological toolbox for TRPV2, a widely expressed membrane protein and orphan drug target. - Nanometric determination of the thickness of aqueous samples for accurate molar absorption coefficients of water-soluble molecules in the mid-infrared region., Gutierrez-Salazar MV & Lorenz-Fonfria VA.
Spectrochim Acta A Mol Biomol Spectrosc (2024)
316: 124378
.
Absorption spectra of aqueous samples measured by transmission need to be acquired using very thin cells (5-50 μm) when targeting the mid-infrared (mid-IR) region due to the strong background absorbance of liquid water. The thickness of the cell used controls the pathlength of the light through the sample, a value needed to transform absorption spectra into molar absorption coefficient spectra, or to determine solute concentrations from absorption spectra. The most accurate way to determine the thickness of an empty cell (i.e., filled with air) is from the period of an interference pattern, known as interference fringes, that arises when the cell is placed perpendicular to the path of light in the spectrometer. However, this same approach is not directly applicable to determine the thickness of a cell filled with an aqueous solution, due partially to the smaller amplitude of the interference fringes but fundamentally caused by its complex waveform, with a wavenumber-dependent oscillation period. Here, using Fresnel equations, we derived analytical expressions to model interference fringes in absorption spectra obtained by transmission, which are also valid for aqueous samples. We also present a novel Fourier-based analysis of the interference fringes that, in combination with the derived analytical expressions, allowed us to determine the pathlength of aqueous samples with an error below ∼ 50 nm. We implemented this novel approach to analyze interference fringes as a Live Script running in the software Matlab. As an application, we measured the absorption spectra of a 97 mM solution of MES buffer at pH 3.4 and pH 8.4 using cells of various nominal thicknesses (6, 25 and 50 μm), whose actual thicknesses were determined using the present approach. The derived molar absorption coefficient spectrum for both the acidic and basic forms of MES were virtually identical regardless of the cell, indicating that the determined thicknesses were likely very accurate. These results illustrate the utility of the present methodology in obtaining accurate molar absorption coefficient spectra of water-soluble molecules in the mid-IR region. - Genetically encoded non-canonical amino acids reveal asynchronous dark reversion of chromophore, backbone, and side-chains in EL222., Chaudhari AS, Chatterjee A, Domingos CAO, Andrikopoulos PC, Liu Y, Andersson I, Schneider B, Lórenz-Fonfría VA & Fuertes G.
Protein Sci (2023)
32: e4590
.
Photoreceptors containing the light-oxygen-voltage (LOV) domain elicit biological responses upon excitation of their flavin mononucleotide (FMN) chromophore by blue light. The mechanism and kinetics of dark-state recovery are not well understood. Here we incorporated the non-canonical amino acid p-cyanophenylalanine (CNF) by genetic code expansion technology at 45 positions of the bacterial transcription factor EL222. Screening of light-induced changes in infrared (IR) absorption frequency, electric field and hydration of the nitrile groups identified residues CNF31 and CNF35 as reporters of monomer/oligomer and caged/decaged equilibria, respectively. Time-resolved multi-probe UV/visible and IR spectroscopy experiments of the lit-to-dark transition revealed four dynamical events. Predominantly, rearrangements around the A'α helix interface (CNF31 and CNF35) precede FMN-cysteinyl adduct scission, folding of α-helices (amide bands), and relaxation of residue CNF151. This study illustrates the importance of characterizing all parts of a protein and suggests a key role for the N-terminal A'α extension of the LOV domain in controlling EL222 photocycle length. - Sub-Millisecond Photoinduced Dynamics of Free and EL222-Bound FMN by Stimulated Raman and Visible Absorption Spectroscopies., Liu Y, Chaudhari AS, Chatterjee A, Andrikopoulos PC, Picchiotti A, Rebarz M, Kloz M, Lorenz-Fonfria VA, Schneider B & Fuertes G.
Biomolecules (2023)
13
.
Time-resolved femtosecond-stimulated Raman spectroscopy (FSRS) provides valuable information on the structural dynamics of biomolecules. However, FSRS has been applied mainly up to the nanoseconds regime and above 700 cm , which covers only part of the spectrum of biologically relevant time scales and Raman shifts. Here we report on a broadband ( 200-2200 cm ) dual transient visible absorption (visTA)/FSRS set-up that can accommodate time delays from a few femtoseconds to several hundreds of microseconds after illumination with an actinic pump. The extended time scale and wavenumber range allowed us to monitor the complete excited-state dynamics of the biological chromophore flavin mononucleotide (FMN), both free in solution and embedded in two variants of the bacterial light-oxygen-voltage (LOV) photoreceptor EL222. The observed lifetimes and intermediate states (singlet, triplet, and adduct) are in agreement with previous time-resolved infrared spectroscopy experiments. Importantly, we found evidence for additional dynamical events, particularly upon analysis of the low-frequency Raman region below 1000 cm . We show that fs-to-sub-ms visTA/FSRS with a broad wavenumber range is a useful tool to characterize short-lived conformationally excited states in flavoproteins and potentially other light-responsive proteins.
2019-22
- Protein conformational changes and protonation dynamics probed by a single shot using quantum-cascade-laser-based IR spectroscopy., Schubert L, Langner P, Ehrenberg D, Lorenz-Fonfria VA & Heberle J.
J Chem Phys (2022)
156: 204201
.
Mid-IR spectroscopy is a powerful and label-free technique to investigate protein reactions. In this study, we use quantum-cascade-laser-based dual-comb spectroscopy to probe protein conformational changes and protonation events by a single-shot experiment. By using a well-characterized membrane protein, bacteriorhodopsin, we provide a comparison between dual-comb spectroscopy and our homebuilt tunable quantum cascade laser (QCL)-based scanning spectrometer as tools to monitor irreversible reactions with high time resolution. In conclusion, QCL-based infrared spectroscopy is demonstrated to be feasible for tracing functionally relevant protein structural changes and proton translocations by single-shot experiments. Thus, we envisage a bright future for applications of this technology for monitoring the kinetics of irreversible reactions as in (bio-)chemical transformations. - Retinal vibrations in bacteriorhodopsin are mechanically harmonic but electrically anharmonic: Evidence from overtone and combination bands., Lorenz-Fonfria VA, Yagi K, Ito S & Kandori H.
Front Mol Biosci (2021)
8
: 749261
.
Fundamental vibrations of the chromophore in the membrane protein bacteriorhodopsin (BR), a protonated Schiff base retinal, have been studied for decades, both by resonance Raman and by infrared (IR) difference spectroscopy. Such studies started comparing vibrational changes between the initial BR state (all- retinal) and the K intermediate (13- retinal), being later extended to the rest of intermediates. They contributed to our understanding of the proton-pumping mechanism of BR by exploiting the sensitivity of fundamental vibrational transitions of the retinal to its conformation. Here, we report on new bands in the 2,500 to 1,800 cm region of the K-BR difference FT-IR spectrum. We show that the bands between 2,500 and 2,300 cm originate from overtone and combination transitions from C-C stretches of the retinal. We assigned bands below 2,300 cm to the combination of retinal C-C stretches with methyl rocks and with hydrogen-out-of-plane vibrations. Remarkably, experimental C-C overtone bands appeared at roughly twice the wavenumber of their fundamentals, with anharmonic mechanical constants ≤3.5 cm , and in some cases of ∼1 cm . Comparison of combination and fundamental bands indicates that most of the mechanical coupling constants are also very small. Despite the mechanical quasi-harmonicity of the C-C stretches, the area of their overtone bands was only ∼50 to ∼100 times smaller than of their fundamental bands. We concluded that electrical anharmonicity, the second mechanism giving intensity to overtone bands, must be particularly high for the retinal C-C stretches. We corroborated the assignments of negative bands in the K-BR difference FT-IR spectrum by ab initio anharmonic vibrational calculations of all-trans retinal in BR using a quantum-mechanics/molecular mechanics approach, reproducing reasonably well the small experimental anharmonic and coupling mechanical constants. Yet, and in spite accounting for both mechanical and electrical anharmonicities, the intensity of overtone C-C transitions was underestimated by a factor of 4-20, indicating room for improvement in state-of-the-art anharmonic vibrational calculations. The relatively intense overtone and combination bands of the retinal might open the possibility to detect retinal conformational changes too subtle to significantly affect fundamental transitions but leaving a footprint in overtone and combination transitions. - A photoswitchable helical peptide with light-controllable interface/transmembrane topology in lipidic membranes, Gutiérrez-Salazar M, Santamaría-Aranda E, Schaar L, Salgado J, Sampedro D & Lorenz-Fonfria VA.
iScience (2021)
24
: 102771
.
The spontaneous insertion of helical transmembrane (TM) polypeptides into lipid bilayers is driven by three sequential equilibria: solution-to-membrane interface (MI) partition, unstructured-to-helical folding, and MI-to-TM helix insertion. A bottleneck for understanding these three steps is the lack of experimental approaches to perturb membrane-bound hydrophobic polypeptides out of equilibrium rapidly and reversibly. Here, we report on a 24-residues-long hydrophobic α-helical polypeptide, covalently coupled to an azobenzene photoswitch (KCALP-azo), which displays a light-controllable TM/MI equilibrium in hydrated lipid bilayers. FTIR spectroscopy reveals that trans KCALP-azo folds as a TM α-helix (TM topology). After trans-to-cis photoisomerization of the azobenzene moiety with UV light (reversed with blue light), the helical structure of KCALP-azo is maintained, but its helix tilt increased from 32 ± 5° to 79 ± 8°, indication of a reversible TM-to-MI transition. Further analysis indicates that this transition is incomplete, with cis KCALP-azo existing in a 90% TM and 10% MI mixture. - Infrared Difference Spectroscopy of Proteins: From Bands to Bonds., Lorenz-Fonfria VA.
Chem Rev (2020)
120: 3466-3576
.
Infrared difference spectroscopy probes vibrational changes of proteins upon their perturbation. Compared with other spectroscopic methods, it stands out by its sensitivity to the protonation state, H-bonding, and the conformation of different groups in proteins, including the peptide backbone, amino acid side chains, internal water molecules, or cofactors. In particular, the detection of protonation and H-bonding changes in a time-resolved manner, not easily obtained by other techniques, is one of the most successful applications of IR difference spectroscopy. The present review deals with the use of perturbations designed to specifically change the protein between two (or more) functionally relevant states, a strategy often referred to as reaction-induced IR difference spectroscopy. In the first half of this contribution, I review the technique of reaction-induced IR difference spectroscopy of proteins, with special emphasis given to the preparation of suitable samples and their characterization, strategies for the perturbation of proteins, and methodologies for time-resolved measurements (from nanoseconds to minutes). The second half of this contribution focuses on the spectral interpretation. It starts by reviewing how changes in H-bonding, medium polarity, and vibrational coupling affect vibrational frequencies, intensities, and bandwidths. It is followed by band assignments, a crucial aspect mostly performed with the help of isotopic labeling and site-directed mutagenesis, and complemented by integration and interpretation of the results in the context of the studied protein, an aspect increasingly supported by spectral calculations. Selected examples from the literature, predominately but not exclusively from retinal proteins, are used to illustrate the topics covered in this review. - Spontaneous and Stress-Induced Pore Formation in Membranes: Theory, Experiments and Simulations., Cunill-Semanat E & Salgado J.
J Membr Biol (2019)
252: 241-260
.
The large plasticity, dynamics and adaptability of biological membranes allow different modes of intrinsic and inducible permeability. These phenomena are of physiological importance for a number of natural functions related to cell death and can also be manipulated artificially for practical purposes like gene transfer, drug delivery, prevention of infections or anticancer therapy. For these advances to develop in a controllable and specific way, we need a sufficient understanding of the membrane permeability phenomena. Since the formulation of early concepts of pore formation, there has been an enormous effort to describe membrane permeability by using theory, simulations and experiments. A major breakthrough has come recently through theoretical developments that allow building continuous trajectories of pore formation both in the absence and presence of stress conditions. The new model provides a coherent quantitative view of membrane permeabilization, useful to test the impact of known lipid properties, make predictions and postulate specific pore intermediates that can be studied by simulations. For example, this theory predicts unprecedented dependencies of the line tension on the pore radius and on applied lateral tension which explain previous puzzling results. In parallel, important concepts have also come from molecular dynamics simulations, of which the role of water for membrane permeabilization is of special interest. These advances open new challenges and perspectives for future progress in the study of membrane permeability, as experiments and simulations will need to test the theoretical predictions, while theory achieves new refinements that provide a physical ground for observations. - Translocation of enzymes into a mesoporous MOF for enhanced catalytic activity under extreme conditions., Navarro-Sánchez J, Almora-Barrios N, Lerma-Berlanga B, Ruiz-Pernía JJ, Lorenz-Fonfria VA, Tuñón I & Martí-Gastaldo C.
Chem Sci (2019)
10: 4082-4088
.
2017-18
- Vibrational and Molecular Properties of Mg2+ Binding and Ion Selectivity in the Magnesium Channel MgtE., Kimura T, Lorenz-Fonfria VA, Douki S, Motoki H, Ishitani R, Nureki O, Higashi M & Furutani Y.
J Phys Chem B (2018)
122: 9681-9696
.
Magnesium ions (Mg ) are crucial for various biological processes. A bacterial Mg channel, MgtE, tightly regulates the intracellular Mg concentration. Previous X-ray crystal structures showed that MgtE forms a dimeric structure composed of a total of 10 transmembrane α helices forming a central pore, and intracellular soluble domains constituting a Mg sensor. The ion selectivity for Mg over Ca resides at a central cavity in the transmembrane pore of MgtE, involving a conserved aspartate residue (Asp432) from each monomer. Here, we applied ion-exchange-induced difference FTIR spectroscopy to analyze the interactions between MgtE and divalent cations, Mg and Ca . Using site-directed mutagenesis, vibrational bands at 1421 (Mg ), 1407 (Mg ), ∼1440 (Ca ), and 1390 (Ca ) cm were assigned to symmetric carboxylate stretching modes of Asp432, involved in the ion coordination. Conservative modifications of the central cavity by Asp432Glu or Ala417Leu mutations resulted in the disappearance of the Mg -sensitive carboxylate bands, suggesting a highly optimized geometry for accommodating a Mg ion. The dependency of the vibrational changes on Mg and Ca concentrations revealed the presence of a two different classes of binding sites: a high affinity site for Mg ( K ≈ 0.3 mM) with low Ca affinity ( K ≈ 80 mM), and a medium affinity site for Mg ( K ≈ 2 mM) and Ca ( K ≈ 6 mM), tentatively assigned to the central cavity and the sensor domain, respectively. With the aid of molecular dynamics simulation and normal-mode analysis by quantum chemistry, we confirm that changes in carboxylate bands of the high affinity binding site originate from Asp432 in the central cavity. - Orientation of non-spherical protonated water clusters revealed by infrared absorption dichroism, Daldrop JO, Saita M, Heyden M, Lórenz-Fonfría VA, Heberle J & Netz RR.
Nat Commun (2018)
9: 311
.
Infrared continuum bands that extend over a broad frequency range are a key spectral signature of protonated water clusters. They are observed for many membrane proteins that contain internal water molecules, but their microscopic mechanism has remained unclear. Here we compute infrared spectra for protonated and unprotonated water chains, discs, and droplets from ab initio molecular dynamics simulations. The continuum bands of the protonated clusters exhibit significant anisotropy for chains and discs, with increased absorption along the direction of maximal cluster extension. We show that the continuum band arises from the nuclei motion near the excess charge, with a long-ranged amplification due to the electronic polarizability. Our experimental, polarization-resolved light–dark difference spectrum of the light-driven proton pump bacteriorhodopsin exhibits a pronounced dichroic continuum band. Our results suggest that the protonated water cluster responsible for the continuum band of bacteriorhodopsin is oriented perpendicularly to the membrane normal. - EXPRESS: Potential Second-Harmonic Ghost Bands in the Fourier Transform Infrared (FT-IR) Difference Spectroscopy of Proteins, Ito S, Kandori H & Lórenz-Fonfría VA.
Appl Spectrosc (2018)
72: 956-963
.
Fourier transform infrared (FT-IR) difference absorption spectroscopy is a common method to study structural and dynamical aspects behind protein function. In particular, the 2800-1800 cm⁻¹ spectral range has been used to obtain information about internal (deuterated) water molecules, as well as site-specific details about cysteine residues and chemically modified and artificial amino acids. Here, we report on the presence of ghost bands in cryogenic light-induced FT-IR difference spectra of the protein bacteriorhodopsin. The presence of these ghost bands can be particularly problematic in the 2800-1900 cm⁻¹ region, showing intensities similar to O-D vibrations from water molecules. We demons trate that they arise from second harmonics from genuine chromophore bands located in the 1400-850 cm⁻¹ region, generated by double-modulation artifacts caused from reflections of the IR beam at the sample and at the cryostat windows back to the interferom eter (inter-reflections). The second-harmonic ghost bands can be physically removed by placing an optical filter of suitable cutoff in the beam path, but at the cost of losing part of the multiplexing advantage of FT-IR spectroscopy. We explored alternativ es to the use of optical filters. Tilting the cryostat windows was effective in reducing the intensity of the second harmonic artifacts but tilting the sample windows was not, presumably by their close proximity to the focal point of the IR beam. We also i ntroduce a simple numerical post-processing approach that can partially, but not fully, correct for second-harmonic ghost bands in FT-IR difference spectra. - Photoexcitation of the P4480 State Induces a Secondary Photocycle That Potentially Desensitizes Channelrhodopsin-2, Saita M, Pranga-Sellnau F, Resler T, Schlesinger R, Heberle J & Lorenz-Fonfria VA.
J Am Chem Soc (2018)
140: 9899-9903
.
Channelrhodopsins (ChRs) are light-gated cation channels. In spite of their wide use to activate neurons with light, the photocurrents of ChRs rapidly decay in intensity under both continuous illumination and fast trains of light pulses, broadly referred to as desensitization. This undesirable phenomenon has been explained by two interconnected photocycles, each of them containing a nonconductive dark state (D1 and D2) and a conductive state (O1 and O2). While the D1 and O1 states correspond to the dark-state and P3520 intermediate of the primary all- trans photocycle of ChR2, the molecular identity of D2 and O2 remains unclear. We show that P4480, the last intermediate of the all- trans photocycle, is photoactive. Its photocycle, characterized by time-resolved UV/vis spectroscopy, contains a red-shifted intermediate, I3530. Our results indicate that the D2 and O2 states correspond to the P4480 and I3530 intermediates, connecting desensitization of ChR2 with the photochemical properties of the P4480 intermediate. - Protein dynamics observed by tunable mid-IR quantum cascade lasers across the time range from 10ns to 1s, Schultz B-J, Mohrmann H, Lórenz-Fonfría VA & Heberle J.
Spectrochim Acta A Mol Biomol Spectrosc (2018)
188: 666-674
.
We have developed a spectrometer based on tunable quantum cascade lasers (QCLs) for recording time-resolved absorption spectra of proteins in the mid-infrared range. We illustrate its performance by recording time-resolved difference spectra of bacteriorhodopsin in the carboxylic range (1800-1700cm-1) and on the CO rebinding reaction of myoglobin (1960-1840cm-1), at a spectral resolution of 1cm-1. The spectrometric setup covers the time range from 4ns to nearly a second with a response time of 10-15ns. Absorption changes as low as 1×10-4 are detected in single-shot experiments at t>1μs, and of 5×10-6 in kinetics obtained after averaging 100 shots. While previous time-resolved IR experiments have mostly been conducted on hydrated films of proteins, we demonstrate here that the brilliance of tunable quantum cascade lasers is superior to perform ns time-resolved experiments even in aqueous solution (H2O) - The Grateful Infrared: Sequential Protein Structural Changes Resolved by Infrared Difference Spectroscopy, Kottke T, Lórenz-Fonfría VA & Heberle J.
J Phys Chem B (2017)
121: 335-350
.
The catalytic activity of proteins is a function of structural changes. Very often these are as minute as protonation changes, hydrogen bonding changes, and amino acid side chain reorientations. To resolve these, a methodology is afforded that not only provides the molecular sensitivity but allows for tracing the sequence of these hierarchical reactions at the same time. This feature article showcases results from time-resolved IR spectroscopy on channelrhodopsin (ChR), light-oxygen-voltage (LOV) domain protein, and cryptochrome (CRY). All three proteins are activated by blue light, but their biological role is drastically different. Channelrhodopsin is a transmembrane retinylidene protein which represents the first light-activated ion channel of its kind and which is involved in primitive vision (phototaxis) of algae. LOV and CRY are flavin-binding proteins acting as photoreceptors in a variety of signal transduction mechanisms in all kingdoms of life. Beyond their biological relevance, these proteins are employed in exciting optogenetic applications. We show here how IR difference absorption resolves crucial structural changes of the protein after photonic activation of the chromophore. Time-resolved techniques are introduced that cover the time range from nanoseconds to minutes along with some technical considerations. Finally, we provide an outlook toward novel experimental approaches that are currently developed in our laboratories or are just in our minds ('Gedankenexperimente'). We believe that some of them have the potential to provide new science - pH-sensitive vibrational probe reveals a cytoplasmic protonated cluster in bacteriorhodopsin, Lorenz-Fonfria VA, Saita M, Lazarova T, Schlesinger R & Heberle J.
Proc Natl Acad Sci U S A (2017)
114: E10909-E10918
.
Infrared spectroscopy has been used in the past to probe the dynamics of internal proton transfer reactions taking place during the functional mechanism of proteins but has remained mostly silent to protonation changes in the aqueous medium. Here, by selectively monitoring vibrational changes of buffer molecules with a temporal resolution of 6 µs, we have traced proton release and uptake events in the light-driven proton-pump bacteriorhodopsin and correlate these to other molecular processes within the protein. We demonstrate that two distinct chemical entities contribute to the temporal evolution and spectral shape of the continuum band, an unusually broad band extending from 2,300 to well below 1,700 cm-1 The first contribution corresponds to deprotonation of the proton release complex (PRC), a complex in the extracellular domain of bacteriorhodopsin where an excess proton is shared by a cluster of internal water molecules and/or ionic E194/E204 carboxylic groups. We assign the second component of the continuum band to the proton uptake complex, a cluster with an excess proton reminiscent to the PRC but located in the cytoplasmic domain and possibly stabilized by D38. Our findings refine the current interpretation of the continuum band and call for a reevaluation of the last proton transfer steps in bacteriorhodopsin
2015-16
- Lysyl oxidase-like 2 (LOXL2) oxidizes trimethylated lysine 4 in histone H3, Herranz N, Dave N, Millanes-Romero A, Pascual-Reguant L, Morey L, Díaz VM, Lórenz-Fonfría V, Gutierrez-Gallego R, Jerónimo C, Iturbide A, Croce LD, de Herreros AG & Peiró S.
FEBS J (2016)
283: 4263-4273.
Methylation of histone H3 lysine 4 is linked to active transcription and can be removed by LSD1 or the JmjC domain-containing proteins by amino-oxidation or hydroxylation, respectively. Here we describe that its deamination can be catalyzed by lysyl oxidase-like 2 protein (LOXL2), presenting an unconventional chemical mechanism for H3K4 modification. Infrared spectroscopy and mass spectrometry analyses demonstrated that recombinant LOXL2 specifically deaminates trimethylated H3K4. Moreover, by regulating H3K4me3 deamination, LOXL2 activity is linked with the transcriptional control of the CDH1 gene. These results reveal the existence of further H3 modification as well as a novel mechanism for H3K4me3 demethylation. - Transient Conformational Changes of Sensory Rhodopsin II Investigated by Vibrational Stark Effect Probes, Mohrmann H, Kube I, Lórenz-Fonfría VA, Engelhard M & Heberle J.
J Phys Chem B (2016)
120: 4383-4387.
Sensory rhodopsin II (SRII) is the primary light sensor in the photophobic reaction of the halobacterium Natronomonas pharaonis. Photoactivation of SRII results in a movement of helices F and G of this seven-helical transmembrane protein. This conformational change is conveyed to the transducer protein (HtrII). Global changes in the protein backbone have been monitored by IR difference spectroscopy by recording frequency shifts in the amide bands. Here we investigate local structural changes by judiciously inserting thiocyanides at different locations of SRII. These vibrational Stark probes absorb in a frequency range devoid of any protein vibrations and respond to local changes in the dielectric, electrostatics, and hydrogen bonding. As a proof of principle, we demonstrate the use of Stark probes to test the conformational changes occurring in SRII 12 ms after photoexcitation and later. Thus, a methodology is provided to trace local conformational changes in membrane proteins by a minimal invasive probe at the high temporal resolution inherent to IR spectroscopy - Overlap and diversity in antimicrobial peptide databases: compiling a non-redundant set of sequences, Aguilera-Mendoza L, Marrero-Ponce Y, Tellez-Ibarra R, Llorente-Quesada MT, Salgado J, Barigye SJ & Liu J.
Bioinformatics (2015)
31: 2553-2559.
The large variety of antimicrobial peptide (AMP) databases developed to date are characterized by a substantial overlap of data and similarity of sequences. Our goals are to analyze the levels of redundancy for all available AMP databases and use this information to build a new non-redundant sequence database. For this purpose, a new software tool is introduced. A comparative study of 25 AMP databases reveals the overlap and diversity among them and the internal diversity within each database. The overlap analysis shows that only one database (Peptaibol) contains exclusive data, not present in any other, whereas all sequences in the LAMP_Patent database are included in CAMP_Patent. However, the majority of databases have their own set of unique sequences, as well as some overlap with other databases. The complete set of non-duplicate sequences comprises 16 990 cases, which is almost half of the total number of reported peptides. On the other hand, the diversity analysis identifies the most and least diverse databases and proves that all databases exhibit some level of redundancy. Finally, we present a new parallel-free software, named Dover Analyzer, developed to compute the overlap and diversity between any number of databases and compile a set of non-redundant sequences. These results are useful for selecting or building a suitable representative set of AMPs, according to specific needs - The melibiose transporter of escherichia coli: Critical contribution of Lys-377 to the structural organization of the interacting substrate binding sites, Fuerst O, Lin Y, Granell M, Leblanc G, Padrós E, Lórenz-Fonfría VA & Cladera J.
J Biol Chem (2015)
290: 16261-16271.
We examine the role of Lys-377, the only charged residue in helix XI, on the functional mechanism of the Na(+)-sugar melibiose symporter from Escherichia coli. Intrinsic fluorescence, FRET, and Fourier transform infrared difference spectroscopy reveal that replacement of Lys-377 with either Cys, Val, Arg, or Asp disables both Na(+) and melibiose binding. On the other hand, molecular dynamics simulations extending up to 200-330 ns reveal that Lys-377 (helix XI) interacts with the anionic side chains of two of the three putative ligands for cation binding (Asp-55 and Asp-59 in helix II). When Asp-59 is protonated during the simulations, Lys-377 preferentially interacts with Asp-55. Interestingly, when a Na(+) ion is positioned in the Asp-55-Asp-59 environment, Asp-124 in helix IV (a residue essential for melibiose binding) reorients and approximates the Asp-55-Asp-59 pair, and all three acidic side chains act as Na(+) ligands. Under these conditions, the side chain of Lys-377 interacts with the carboxylic moiety of these three Asp residues. These data highlight the crucial role of the Lys-377 residue in the spatial organization of the Na(+) binding site. Finally, the analysis of the second-site revertants of K377C reveals that mutation of Ile-22 (in helix I) preserves Na(+) binding, whereas that of melibiose is largely abolished according to spectroscopic measurements. This amino acid is located in the border of the sugar-binding site and might participate in sugar binding through apolar interactions - Direct observation of nanometer-scale pores of melittin in supported lipid monolayers, Giménez D, Sánchez-Muñoz OL & Salgado J.
Langmuir (2015)
31: 3146-3158.
Melittin is the most studied membrane-active peptide and archetype within a large and diverse group of pore formers. However, the molecular characteristics of melittin pores remain largely unknown. Herein, we show by atomic force microscopy (AFM) that lipid monolayers in the presence of melittin are decorated with numerous regularly shaped circular pores that can be distinguished from nonspecific monolayer defects. The specificity of these pores is reinforced through a statistical evaluation of depressions found in Langmuir-Blodgett monolayers in the presence and absence of melittin, which eventually allows characterization of the melittin-induced pores at a quantitative low-resolution level. We observed that the large majority of pores exhibit near-circular symmetry and a Gaussian distribution in size, with a mean diameter of ∼8.7 nm. A distinctive feature is a ring of material found around the pores, made by, on average, three positive peaks, with a height over the level of the lipidic background of ∼0.23 nm. This protruding rim is most likely due to the presence of melittin near the pore border. Although the current resolution of the AFM images in the x, y plane does not allow distinction of the specific organization of the peptide molecules, these results provide an unprecedented view of melittin pores formed in lipidic interfaces and open new perspectives for future structural investigations of these and other pore-forming peptides and proteins using supported monolayers - Pre-gating conformational changes in the ChETA variant of channelrhodopsin-2 monitored by nanosecond IR spectroscopy, Lórenz-Fonfría VA, Schultz B-J, Resler T, Schlesinger R, Bamann C, Bamberg E & Heberle J.
J Am Chem Soc (2015)
137: 1850-1861.
Light-gated ion permeation by channelrhodopsin-2 (ChR2) relies on the photoisomerization of the retinal chromophore and the subsequent photocycle, leading to the formation (on-gating) and decay (off-gating) of the conductive state. Here, we have analyzed the photocycle of a fast-cycling ChR2 variant (E123T mutation, also known as ChETA), by time-resolved UV/vis, step-scan FT-IR, and tunable quantum cascade laser IR spectroscopies with nanosecond resolution. Pre-gating conformational changes rise with a half-life of 200 ns, silent to UV/vis but detected by IR spectroscopy. They involve changes in the peptide backbone and in the H-bond of the side chain of the critical residue D156. Thus, the P1(500) intermediate must be separated into early and late states. Light-adapted ChR2 contains a mixture of all-trans and 13-cis retinal in a 70:30 ratio which are both photoactive. Analysis of ethylenic and fingerprint vibrations of retinal provides evidence that the 13-cis photocycle recovers in 1 ms. This recovery is faster than channel off-gating and most of the proton transfer reactions, implying that the 13-cis photocycle is of minor functional relevance for ChR2 - Temporal evolution of helix hydration in a light-gated ion channel correlates with ion conductance, Lórenz-Fonfría VA, Bamann C, Resler T, Schlesinger R, Bamberg E & Heberle J.
Proc Natl Acad Sci U S A (2015)
112: E5796-E5804.
The discovery of channelrhodopsins introduced a new class of light-gated ion channels, which when genetically encoded in host cells resulted in the development of optogenetics. Channelrhodopsin-2 from Chlamydomonas reinhardtii, CrChR2, is the most widely used optogenetic tool in neuroscience. To explore the connection between the gating mechanism and the influx and efflux of water molecules in CrChR2, we have integrated light-induced time-resolved infrared spectroscopy and electrophysiology. Cross-correlation analysis revealed that ion conductance tallies with peptide backbone amide I vibrational changes at 1,665(-) and 1,648(+) cm(-1). These two bands report on the hydration of transmembrane α-helices as concluded from vibrational coupling experiments. Lifetime distribution analysis shows that water influx proceeded in two temporally separated steps with time constants of 10 μs (30%) and 200 μs (70%), the latter phase concurrent with the start of ion conductance. Water efflux and the cessation of the ion conductance are synchronized as well, with a time constant of 10 ms. The temporal correlation between ion conductance and hydration of helices holds for fast (E123T) and slow (D156E) variants of CrChR2, strengthening its functional significance - Kinetic and vibrational isotope effects of proton transfer reactions in channelrhodopsin-2, Resler T, Schultz B-J, Lórenz-Fonfría VA, Schlesinger R & Heberle J.
Biophys J (2015)
109: 287-297.
Channelrhodopsins (ChRs) are light-gated cation channels. After blue-light excitation, the protein undergoes a photocycle with different intermediates. Here, we have recorded transient absorbance changes of ChR2 from Chlamydomonas reinhardtii in the visible and infrared regions with nanosecond time resolution, the latter being accomplished using tunable quantum cascade lasers. Because proton transfer reactions play a key role in channel gating, we determined vibrational as well as kinetic isotope effects (VIEs and KIEs) of carboxylic groups of various key aspartic and glutamic acid residues by monitoring their C=O stretching vibrations in H2O and in D2O. D156 exhibits a substantial KIE (>2) in its deprotonation and reprotonation, which substantiates its role as the internal proton donor to the retinal Schiff base. The unusual VIE of D156, upshifted from 1736 cm(-1) to 1738 cm(-1) in D2O, was scrutinized by studying the D156E variant. The C=O stretch of E156 shifted down by 8 cm(-1) in D2O, providing evidence for the accessibility of the carboxylic group. The C=O stretching band of E90 exhibits a VIE of 9 cm(-1) and a KIE of ∼2 for the de- and the reprotonation reactions during the lifetime of the late desensitized state. The KIE of 1 determined in the time range from 20 ns to 5 ms is incompatible with early deprotonation of E90 - A Hooke׳s law-based approach to protein folding rate, Ruiz-Blanco YB, Marrero-Ponce Y, Prieto PJ, Salgado J, García Y & Sotomayor-Torres CM.
J Theor Biol (2015)
364: 407-417.
Kinetics is a key aspect of the renowned protein folding problem. Here, we propose a comprehensive approach to folding kinetics where a polypeptide chain is assumed to behave as an elastic material described by the Hooke׳s law. A novel parameter called elastic-folding constant results from our model and is suggested to distinguish between protein with two-state and multi-state folding pathways. A contact-free descriptor, named folding degree, is introduced as a suitable structural feature to study protein-folding kinetics. This approach generalizes the observed correlations between varieties of structural descriptors with the folding rate constant. Additionally several comparisons among structural classes and folding mechanisms were carried out showing the good performance of our model with proteins of different types. The present model constitutes a simple rationale for the structural and energetic factors involved in protein folding kinetics
2013-14
- (19)F NMR screening of unrelated antimicrobial peptides shows that membrane interactions are largely governed by lipids, Afonin S, Glaser RW, Sachse C, Salgado J, Wadhwani P & Ulrich AS.
Biochim Biophys Acta, Biomembr (2014)
1838: 2260-2268.
Many amphiphilic antimicrobial peptides permeabilize bacterial membranes via successive steps of binding, re-alignment and/or oligomerization. Here, we have systematically compared the lipid interactions of two structurally unrelated peptides: the cyclic β-pleated gramicidin S (GS), and the α-helical PGLa. (19)F NMR was used to screen their molecular alignment in various model membranes over a wide range of temperatures. Both peptides were found to respond to the phase state and composition of these different samples in a similar way. In phosphatidylcholines, both peptides first bind to the bilayer surface. Above a certain threshold concentration they can re-align and immerse more deeply into the hydrophobic core, which presumably involves oligomerization. Re-alignment is most favorable around the lipid chain melting temperature, and also promoted by decreasing bilayer thickness. The presence of anionic lipids has no influence in fluid membranes, but in the gel phase the alignment states are more complex. Unsaturated acyl chains and other lipids with intrinsic negative curvature prevent re-alignment, hence GS and PGLa do not insert into mixtures resembling bacterial membranes, nor into bacterial lipid extracts. Cholesterol, which is present in high concentrations in animal membranes, even leads to an expulsion of the peptides from the bilayer and prevents their binding altogether. However, a very low cholesterol content of 10% was found to promote binding and re-alignment of both peptides. Overall, these findings show that the ability of amphiphilic peptides to re-align and immerse into a membrane is determined by the physico-chemical properties of the lipids, such as spontaneous curvature. This idea is reinforced by the remarkably similar behavior observed here for two structurally unrelated molecules (with different conformation, size, shape, charge), which further suggests that their activity at the membrane level is largely governed by the properties of the constituent lipids, while the selectivity towards different cell types is additionally ruled by electrostatic attraction between peptide and cell surface. This article is part of a Special Issue entitled: Interfacially Active Peptides and Proteins. Guest Editors: William C. Wimley and Kalina Hristova - Channelrhodopsin unchained: structure and mechanism of a light-gated cation channel, Lórenz-Fonfría VA & Heberle J.
Biochim Biophys Acta, Bioenerg (2014)
1837: 626-642.
The new and vibrant field of optogenetics was founded by the seminal discovery of channelrhodopsin, the first light-gated cation channel. Despite the numerous applications that have revolutionised neurophysiology, the functional mechanism is far from understood on the molecular level. An arsenal of biophysical techniques has been established in the last decades of research on microbial rhodopsins. However, application of these techniques is hampered by the duration and the complexity of the photoreaction of channelrhodopsin compared with other microbial rhodopsins. A particular interest in resolving the molecular mechanism lies in the structural changes that lead to channel opening and closure. Here, we review the current structural and mechanistic knowledge that has been accomplished by integrating the static structure provided by X-ray crystallography and electron microscopy with time-resolved spectroscopic and electrophysiological techniques. The dynamical reactions of the chromophore are effectively coupled to structural changes of the protein, as shown by ultrafast spectroscopy. The hierarchical sequence of structural changes in the protein backbone that spans the time range from 10(-12)s to 10(-3)s prepares the channel to open and, consequently, cations can pass. Proton transfer reactions that are associated with channel gating have been resolved. In particular, glutamate 253 and aspartic acid 156 were identified as proton acceptor and donor to the retinal Schiff base. The reprotonation of the latter is the critical determinant for channel closure. The proton pathway that eventually leads to proton pumping is also discussed. This article is part of a Special Issue entitled: Retinal Proteins - You can teach an old dog new tricks - Changes in the hydrogen-bonding strength of internal water molecules and cysteine residues in the conductive state of channelrhodopsin-1, Lórenz-Fonfría VA, Muders V, Schlesinger R & Heberle J.
J Chem Phys (2014)
141: 22D507.
Water plays an essential role in the structure and function of proteins, particularly in the less understood class of membrane proteins. As the first of its kind, channelrhodopsin is a light-gated cation channel and paved the way for the new and vibrant field of optogenetics, where nerve cells are activated by light. Still, the molecular mechanism of channelrhodopsin is not understood. Here, we applied time-resolved FT-IR difference spectroscopy to channelrhodopsin-1 from Chlamydomonas augustae. It is shown that the (conductive) P2(380) intermediate decays with τ ≈ 40 ms and 200 ms after pulsed excitation. The vibrational changes between the closed and the conductive states were analyzed in the X-H stretching region (X = O, S, N), comprising vibrational changes of water molecules, sulfhydryl groups of cysteine side chains and changes of the amide A of the protein backbone. The O-H stretching vibrations of 'dangling' water molecules were detected in two different states of the protein using H2 (18)O exchange. Uncoupling experiments with a 1:1 mixture of H2O:D2O provided the natural uncoupled frequencies of the four O-H (and O-D) stretches of these water molecules, each with a very weakly hydrogen-bonded O-H group (3639 and 3628 cm(-1)) and with the other O-H group medium (3440 cm(-1)) to moderately strongly (3300 cm(-1)) hydrogen-bonded. Changes in amide A and thiol vibrations report on global and local changes, respectively, associated with the formation of the conductive state. Future studies will aim at assigning the respective cysteine group(s) and at localizing the 'dangling' water molecules within the protein, providing a better understanding of their functional relevance in CaChR1 - Proton transfer and protein conformation dynamics in photosensitive proteins by time-resolved step-scan Fourier-transform infrared spectrosc, Lórenz-Fonfría VA & Heberle J.
J Vis Exp (2014)
: e51622.
Monitoring the dynamics of protonation and protein backbone conformation changes during the function of a protein is an essential step towards understanding its mechanism. Protonation and conformational changes affect the vibration pattern of amino acid side chains and of the peptide bond, respectively, both of which can be probed by infrared (IR) difference spectroscopy. For proteins whose function can be repetitively and reproducibly triggered by light, it is possible to obtain infrared difference spectra with (sub)microsecond resolution over a broad spectral range using the step-scan Fourier transform infrared technique. With -10(2)-10(3) repetitions of the photoreaction, the minimum number to complete a scan at reasonable spectral resolution and bandwidth, the noise level in the absorption difference spectra can be as low as -10(-) (4), sufficient to follow the kinetics of protonation changes from a single amino acid. Lower noise levels can be accomplished by more data averaging and/or mathematical processing. The amount of protein required for optimal results is between 5-100 µg, depending on the sampling technique used. Regarding additional requirements, the protein needs to be first concentrated in a low ionic strength buffer and then dried to form a film. The protein film is hydrated prior to the experiment, either with little droplets of water or under controlled atmospheric humidity. The attained hydration level (g of water / g of protein) is gauged from an IR absorption spectrum. To showcase the technique, we studied the photocycle of the light-driven proton-pump bacteriorhodopsin in its native purple membrane environment, and of the light-gated ion channel channelrhodopsin-2 solubilized in detergent - Resonance Raman and FTIR spectroscopic characterization of the closed and open states of channelrhodopsin-1, Muders V, Kerruth S, Lórenz-Fonfría VA, Bamann C, Heberle J & Schlesinger R.
FEBS Lett (2014)
588: 2301-2306.
Channelrhodopsin-1 from Chlamydomonas augustae (CaChR1) is a light-activated cation channel, which is a promising optogenetic tool. We show by resonance Raman spectroscopy and retinal extraction followed by high pressure liquid chromatography (HPLC) that the isomeric ratio of all-trans to 13-cis of solubilized channelrhodopsin-1 is with 70:30 identical to channelrhodopsin-2 from Chlamydomonas reinhardtii (CrChR2). Critical frequency shifts in the retinal vibrations are identified in the Raman spectrum upon transition to the open (conductive P2(380)) state. Fourier transform infrared spectroscopy (FTIR) spectra indicate different structures of the open states in the two channelrhodopsins as reflected by the amide I bands and the protonation pattern of acidic amino acids - Reaction monitoring using mid-infrared laser-based vibrational circular dichroism, Rüther A, Pfeifer M, Lórenz-Fonfría VA & Lüdeke S.
Chirality (2014)
26: 490-496.
Changes in vibrational circular dichroism (VCD) were recorded on-line during a chemical reaction. The chiral complex nickel-(-)-sparteine chloride was hydrolyzed to free (-)-sparteine base in a biphasic system of sodium hydroxide solution and chloroform (CHCl(3)). Infrared (IR) and VCD spectra were iteratively recorded after pumping a sample from the CHCl(3) phase through a lab-built VCD spectrometer equipped with a tunable mid-IR quantum cascade laser light source, which allows for VCD measurements even in the presence of strongly absorbing backgrounds. Time-dependent VCD spectra were analyzed by singular value decomposition and global exponential fitting. Spectral features corresponding to the complex and free (-)-sparteine could be clearly identified in the fitted amplitude spectrum, which was associated with an exponential decay with an apparent time constant of 127 min (t(½) = 88 min) - pH titration monitored by quantum cascade laser-based vibrational circular dichroism, Rüther A, Pfeifer M, Lórenz-Fonfría VA & Lüdeke S.
J Phys Chem B (2014)
118: 3941-3949.
Vibrational circular dichroism (VCD) spectra of aqueous solutions of proline were recorded in the course of titrations from basic to acidic pH using a spectrometer equipped with a quantum cascade laser (QCL) as an infrared light source in the spectral range from 1320 to 1220 cm(-1). The pH-dependent spectra were analyzed by singular value decomposition and global fitting of a two-pK Henderson-Hasselbalch model. The analysis delivered relative fractions of the three different protonation species. Their agreement with the relative fractions obtained from performing the same analysis on pH-dependent Fourier transform infrared (FT-IR) and QCL-IR spectra validates the quantitative results from QCL-VCD. Global fitting of the pH-dependent VCD spectra of L-proline allowed for extraction of pure spectra corresponding to anionic, zwitterionic, and cationic L-proline. From a static experiment, only pure spectra of the zwitterion would be accessible in a straightforward way. A comparison to VCD spectra calculated for all three species led to assignment of vibrational modes that are characteristic for the respective protonation states. The study demonstrates the applicability of QCL-VCD both for quantitative evaluation and for qualitative interpretation of dynamic processes in aqueous solutions - The substitution of Arg149 with Cys fixes the melibiose transporter in an inward-open conformation, Lin Y, Fuerst O, Granell M, Leblanc G, Lórenz-Fonfría V & Padrós E.
Biochim Biophys Acta, Biomembr (2013)
1828: 1690-1699.
The melibiose transporter from Escherichia coli (MelB) can use the electrochemical energy of either H+, Na+ or Li+ to transport the disaccharide melibiose to the cell interior. By using spectroscopic and biochemical methods, we have analyzed the role of Arg149 by mutagenesis. According to Fourier transform infrared difference and fluorescence spectroscopy studies, R149C, R149Q and R149K all bind substrates in proteoliposomes, where the protein is disposed inside-out. Analysis of right-side-out (RSO) and inside-out (ISO) membrane vesicles showed that the functionally active R149Q and R149K mutants could bind externally added fluorescent sugar analog in both types of vesicles. In contrast, the non-transporting R149C mutant does bind the fluorescent sugar analog as well as melibiose and Na+ in ISO, but not in RSO vesicles. Therefore, the mutation of Arg149 into cysteine restrains the orientation of transporter to an inward-open conformation, with the inherent consequences of a) reducing the frequency of access of outer substrates to the binding sites, and b) impairing active transport. It is concluded that Arg149, most likely located in the inner (cytoplasmic) half of transmembrane helix 5, is critically involved in the reorientation mechanism of the substrate-binding site accessibility in MelB. - Transient protonation changes in channelrhodopsin-2 and their relevance to channel gating, Lórenz-Fonfría VA, Resler T, Krause N, Nack M, Gossing M, Fischer von Mollard G, Bamann C, Bamberg E, Schlesinger R & Heberle J.
Proc Natl Acad Sci U S A (2013)
110: E1273-E1281.
The discovery of the light-gated ion channel channelrhodopsin (ChR) set the stage for the novel field of optogenetics, where cellular processes are controlled by light. However, the underlying molecular mechanism of light-induced cation permeation in ChR2 remains unknown. Here, we have traced the structural changes of ChR2 by time-resolved FTIR spectroscopy, complemented by functional electrophysiological measurements. We have resolved the vibrational changes associated with the open states of the channel (P(2)(390) and P(3)(520)) and characterized several proton transfer events. Analysis of the amide I vibrations suggests a transient increase in hydration of transmembrane α-helices with a t(1/2) = 60 μs, which tallies with the onset of cation permeation. Aspartate 253 accepts the proton released by the Schiff base (t(1/2) = 10 μs), with the latter being reprotonated by aspartic acid 156 (t(1/2) = 2 ms). The internal proton acceptor and donor groups, corresponding to D212 and D115 in bacteriorhodopsin, are clearly different from other microbial rhodopsins, indicating that their spatial position in the protein was relocated during evolution. Previous conclusions on the involvement of glutamic acid 90 in channel opening are ruled out by demonstrating that E90 deprotonates exclusively in the nonconductive P(4)(480) state. Our results merge into a mechanistic proposal that relates the observed proton transfer reactions and the protein conformational changes to the gating of the cation channel - Global stability of protein folding from an empirical free energy function, Ruiz-Blanco YB, Marrero-Ponce Y, Paz W, García Y & Salgado J.
J Theor Biol (2013)
321: 44-53.
The principles governing protein folding stand as one of the biggest challenges of Biophysics. Modeling the global stability of proteins and predicting their tertiary structure are hard tasks, due in part to the variety and large number of forces involved and the difficulties to describe them with sufficient accuracy. We have developed a fast, physics-based empirical potential, intended to be used in global structure prediction methods. This model considers four main contributions: Two entropic factors, the hydrophobic effect and configurational entropy, and two terms resulting from a decomposition of close-packing interactions, namely the balance of the dispersive interactions of folded and unfolded states and electrostatic interactions between residues. The parameters of the model were fixed from a protein data set whose unfolding free energy has been measured at the 'standard' experimental conditions proposed by Maxwell et al. (2005) and a large data set of 1151 monomeric proteins obtained from the PDB. A blind test with proteins taken from ProTherm database, at similar experimental conditions, was carried out. We found a good correlation with the test data set, proving the effectiveness of our model for predicting protein folding free energies in considered standard conditions. Such a prediction compares favorably against estimations made with FoldX's function and the force field GROMOS96. This model constitutes a valuable tool for the fast evaluation of protein structure stability in 3D structure prediction methods - Canonical azimuthal rotations and flanking residues constrain the orientation of transmembrane helices, Sánchez-Muñoz OL, Strandberg E, Esteban-Martín S, Grage SL, Ulrich AS & Salgado J.
Biophys J (2013)
104: 1508-1516.
In biological membranes the alignment of embedded proteins provides crucial structural information. The transmembrane (TM) parts have well-defined secondary structures, in most cases α-helices and their orientation is given by a tilt angle and an azimuthal rotation angle around the main axis. The tilt angle is readily visualized and has been found to be functionally relevant. However, there exist no general concepts on the corresponding azimuthal rotation. Here, we show that TM helices prefer discrete rotation angles. They arise from a combination of intrinsic properties of the helix geometry plus the influence of the position and type of flanking residues at both ends of the hydrophobic core. The helical geometry gives rise to canonical azimuthal angles for which the side chains of residues from the two ends of the TM helix tend to have maximum or minimum immersion within the membrane. This affects the preferential position of residues that fall near hydrophobic/polar interfaces of the membrane, depending on their hydrophobicity and capacity to form specific anchoring interactions. On this basis, we can explain the orientation and dynamics of TM helices and make accurate predictions, which correspond well to the experimental values of several model peptides (including dimers), and TM segments of polytopic membrane proteins
2011-12
- Comparative analysis of the orientation of transmembrane peptides using solid-state (2)H- and (15)N-NMR: mobility matters, Grage SL, Strandberg E, Wadhwani P, Esteban-Martín S, Salgado J & Ulrich AS.
Eur Biophys J (2012)
41: 475-482.
Many solid-state nuclear magnetic resonance (NMR) approaches for membrane proteins rely on orientation-dependent parameters, from which the alignment of peptide segments in the lipid bilayer can be calculated. Molecules embedded in liquid-crystalline membranes, such as monomeric helices, are highly mobile, leading to partial averaging of the measured NMR parameters. These dynamic effects need to be taken into account to avoid misinterpretation of NMR data. Here, we compare two common NMR approaches: (2)H-NMR quadrupolar waves, and separated local field (15)N-(1)H polarization inversion spin exchange at magic angle (PISEMA) spectra, in order to identify their strengths and drawbacks for correctly determining the orientation and mobility of α-helical transmembrane peptides. We first analyzed the model peptide WLP23 in oriented dimyristoylphosphatidylcholine (DMPC) membranes and then contrasted it with published data on GWALP23 in dilauroylphosphatidylcholine (DLPC). We only obtained consistent tilt angles from the two methods when taking dynamics into account. Interestingly, the two related peptides differ fundamentally in their mobility. Although both helices adopt the same tilt in their respective bilayers ( 20°), WLP23 undergoes extensive fluctuations in its azimuthal rotation angle, whereas GWALP23 is much less dynamic. Both alternative NMR methods are suitable for characterizing orientation and dynamics, yet they can be optimally used to address different aspects. PISEMA spectra immediately reveal the presence of large-amplitude rotational fluctuations, which are not directly seen by (2)H-NMR. On the other hand, PISEMA was unable to define the azimuthal rotation angle in the case of the highly dynamic WLP23, though the helix tilt could still be determined, irrespective of any dynamics parameters - Studying substrate binding to reconstituted secondary transporters by attenuated total reflection infrared difference spectroscopy, Lórenz-Fonfría VA, León X & Padrós E.
Methods Mol Biol (2012)
914: 107-126.
The determination of protein conformational changes induced by the interaction of substrates with secondary transporters is an important step toward the elucidation of their transport mechanism. Since conformational changes in a protein alter its vibrational patterns, they can be detected with high sensitivity by infrared difference (IR(diff)) spectroscopy without the need for external probes. We describe a general procedure to obtain substrate-induced IR(diff) spectra by alternating perfusion of buffers over an attenuated total reflection (ATR) crystal containing an adhered film of a membrane protein reconstituted in lipids. As an example, we provide specific protocols to obtain melibiose and Na(+)-induced ATR-IR(diff) spectra of reconstituted melibiose permease, a sodium/melibiose co-transporter from E. coli. The presented methodology is applicable in principle to any membrane protein, provided that it can be purified and reconstituted in functional form, and appropriate substrates are available - Probing a polar cluster in the retinal binding pocket of bacteriorhodopsin by a chemical design approach, Simón-Vázquez R, Domínguez M, Lórenz-Fonfría VA, Alvarez S, Bourdelande J-L, de Lera AR, Padrós E & Perálvarez-Marín A.
PLoS One (2012)
7: e42447.
Bacteriorhodopsin has a polar cluster of amino acids surrounding the retinal molecule, which is responsible for light harvesting to fuel proton pumping. From our previous studies, we have shown that threonine 90 is the pivotal amino acid in this polar cluster, both functionally and structurally. In an attempt to perform a phenotype rescue, we have chemically designed a retinal analogue molecule to compensate the drastic effects of the T90A mutation in bacteriorhodopsin. This analogue substitutes the methyl group at position C(13) of the retinal hydrocarbon chain by and ethyl group (20-methyl retinal). We have analyzed the effect of reconstituting the wild-type and the T90A mutant apoproteins with all-trans-retinal and its 20-methyl derivative (hereafter, 13-ethyl retinal). Biophysical characterization indicates that recovering the steric interaction between the residue 90 and retinal, eases the accommodation of the chromophore, however it is not enough for a complete phenotype rescue. The characterization of these chemically engineered chromoproteins provides further insight into the role of the hydrogen bond network and the steric interactions involving the retinal binding pocket in bacteriorhodopsin and other microbial sensory rhodopsins - Hydrophobic mismatch of mobile transmembrane helices: Merging theory and experiments, Strandberg E, Esteban-Martín S, Ulrich AS & Salgado J.
Biochim Biophys Acta, Biomembr (2012)
1818: 1242-1249.
Hydrophobic mismatch still represents a puzzle for transmembrane peptides, despite the apparent simplicity of this concept and its demonstrated validity in natural membranes. Using a wealth of available experimental ((2))H NMR data, we provide here a comprehensive explanation of the orientation and dynamics of model peptides in lipid bilayers, which shows how they can adapt to membranes of different thickness. The orientational adjustment of transmembrane α-helices can be understood as the result of a competition between the thermodynamically unfavorable lipid repacking associated with peptide tilting and the optimization of peptide/membrane hydrophobic coupling. In the positive mismatch regime (long-peptide/thin-membrane) the helices adapt mainly via changing their tilt angle, as expected from simple geometrical predictions. However, the adaptation mechanism varies with the peptide sequence in the flanking regions, suggesting additional effects that modulate hydrophobic coupling. These originate from re-adjustments of the peptide hydrophobic length and they depend on the hydrophobicity of the flanking region, the strength of interfacial anchoring, the structural flexibility of anchoring side-chains and the presence of alternative anchoring residues - µ-Calpain conversion of antiapoptotic Bfl-1 (BCL2A1) into a prodeath factor reveals two distinct alpha-helices inducing mitochondria-mediated apoptosis, Valero JG, Cornut-Thibaut A, Jugé R, Debaud AL, Giménez D, Gillet G, Bonnefoy-Bérard N, Salgado J, Salles G, Aouacheria A & Kucharczak J.
PLoS One (2012)
7: e38620.
Anti-apoptotic Bfl-1 and pro-apoptotic Bax, two members of the Bcl-2 family sharing a similar structural fold, are classically viewed as antagonist regulators of apoptosis. However, both proteins were reported to be death inducers following cleavage by the cysteine protease µ-calpain. Here we demonstrate that calpain-mediated cleavage of full-length Bfl-1 induces the release of C-terminal membrane active α-helices that are responsible for its conversion into a pro-apoptotic factor. A careful comparison of the different membrane-active regions present in the Bfl-1 truncated fragments with homologous domains of Bax show that helix α5, but not α6, of Bfl-1 induces cell death and cytochrome c release from purified mitochondria through a Bax/Bak-dependent mechanism. In contrast, both helices α5 and α6 of Bax permeabilize mitochondria regardless of the presence of Bax or Bak. Moreover, we provide evidence that the α9 helix of Bfl-1 promotes cytochrome c release and apoptosis through a unique membrane-destabilizing action whereas Bax-α9 does not display such activities. Hence, despite a common 3D-structure, C-terminal toxic domains present on Bfl-1 and Bax function in a dissimilar manner to permeabilize mitochondria and induce apoptosis. These findings provide insights for designing therapeutic approaches that could exploit the cleavage of endogenous Bcl-2 family proteins or the use of Bfl-1/Bax-derived peptides to promote tumor cell clearance - A lipocentric view of peptide-induced pores, Fuertes G, Giménez D, Esteban-Martín S, Sánchez-Muñoz OL & Salgado J.
Eur Biophys J (2011)
40: 399-415.
Although lipid membranes serve as effective sealing barriers for the passage of most polar solutes, nonmediated leakage is not completely improbable. A high activation energy normally keeps unassisted bilayer permeation at a very low frequency, but lipids are able to self-organize as pores even in peptide-free and protein-free membranes. The probability of leakage phenomena increases under conditions such as phase coexistence, external stress or perturbation associated to binding of nonlipidic molecules. Here, we argue that pore formation can be viewed as an intrinsic property of lipid bilayers, with strong similarities in the structure and mechanism between pores formed with participation of peptides, lipidic pores induced by different types of stress, and spontaneous transient bilayer defects driven by thermal fluctuations. Within such a lipocentric framework, amphipathic peptides are best described as pore-inducing rather than pore-forming elements. Active peptides bound to membranes can be understood as a source of internal surface tension which facilitates pore formation by diminishing the high activation energy barrier. This first or immediate action of the peptide has some resemblance to catalysis. However, the presence of membrane-active peptides has the additional effect of displacing the equilibrium towards the pore-open state, which is then maintained over long times, and reducing the size of initial individual pores. Thus, pore-inducing peptides, regardless of their sequence and oligomeric organization, can be assigned a double role of increasing the probability of pore formation in membranes to high levels as well as stabilizing these pores after they appear - Switchable bactericidal effects from novel silica-coated silver nanoparticles mediated by light irradiation, Fuertes G, Sánchez-Muñoz OL, Pedrueza E, Abderrafi K, Salgado J & Jiménez E.
Langmuir (2011)
27: 2826-2833.
Here we report on the triggering of antibacterial activity by a new type of silver nanoparticle coated with porous silica, Ag@silica, irradiated at their surface plasmon resonant frequency. The nanoparticles are able to bind readily to the surface of bacterial cells, although this does not affect bacterial growth since the silica shell largely attenuates the intrinsic toxicity of silver. However, upon simultaneous exposure to light corresponding to the absorption band of the nanoparticles, bacterial death is enhanced selectively on the irradiated zone. Because of the low power density used for the treatments, we discard thermal effects as the cause of cell killing. Instead, we propose that the increase in toxicity is due to the enhanced electromagnetic field in the proximity of the nanoparticles, which indirectly, most likely through induced photochemical reactions, is able to cause cell death - Probing specific molecular processes and intermediates by time-resolved Fourier transform infrared spectroscopy: application to the bacteriorhodopsin photocycle, Lórenz-Fonfría VA, Kandori H & Padrós E.
J Phys Chem B (2011)
115: 7972-7985.
We present a general approach for probing the kinetics of specific molecular processes in proteins by time-resolved Fourier transform infrared (IR) spectroscopy. Using bacteriorhodopsin (bR) as a model we demonstrate that by appropriately monitoring some selected IR bands it is possible obtaining the kinetics of the most important events occurring in the photocycle, namely changes in the chromophore and the protein backbone conformation, and changes in the protonation state of the key residues implicated in the proton transfers. Besides confirming widely accepted views of the bR photocycle, our analysis also sheds light into some disputed issues: the degree of retinal torsion in the L intermediate to respect the ground state; the possibility of a proton transfer from Asp85 to Asp212; the relationship between the protonation/deprotonation of Asp85 and the proton release complex; and the timing of the protein backbone dynamics. By providing a direct way to estimate the kinetics of photocycle intermediates the present approach opens new prospects for a robust quantitative kinetic analysis of the bR photocycle, which could also benefit the study of other proteins involved in photosynthesis, in phototaxis, or in respiratory chains - Solubilization, Purification, and Characterization of Integral Membrane Proteins, Lórenz-Fonfría V, Perálvarez-Marín A, Padrós E & Lazarova T.
In: Production of Membrane Proteins: Strategies for Expression and Isolation, Robinson, A.S.
(Ed.). Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011,
pp. 317-360.
- Bax-derived membrane-active peptides act as potent and direct inducers of apoptosis in cancer cells, Valero JG, Sancey L, Kucharczak J, Guillemin Y, Giménez D, Prudent J, Gillet G, Salgado J, Coll JL & Aouacheria A.
J Cell Sci (2011)
124: 556-564.
Although many cancer cells are primed for apoptosis, they usually develop resistance to cell death at several levels. Permeabilization of the outer mitochondrial membrane, which is mediated by proapoptotic Bcl-2 family members such as Bax, is considered as a point of no return for initiating apoptotic cell death. This crucial role has placed Bcl-2 family proteins as recurrent targets for anticancer drug development. Here, we propose and demonstrate a new concept based on minimal active versions of Bax to induce cell death independently of endogenous Bcl-2 proteins. We show that membrane-active segments of Bax can directly induce the release of mitochondria-residing apoptogenic factors and commit tumor cells promptly and irreversibly to caspase-dependent apoptosis. On this basis, we designed a peptide encompassing part of the Bax pore-forming domain, which can target mitochondria, induce cytochrome c release and trigger caspase-dependent apoptosis. Moreover, this Bax-derived 'poropeptide' produced effective tumor regression after peritumoral injection in a nude mouse xenograft model. Thus, peptides derived from proteins that form pores in the mitochondrial outer membrane represent novel templates for anticancer agents
2009-10
- Solid state NMR analysis of peptides in membranes: Influence of dynamics and labeling scheme, Esteban-Martín S, Strandberg E, Salgado J & Ulrich AS.
Biochim Biophys Acta, Biomembr (2010)
1798: 252-257.
The functional state of a membrane-active peptide is often defined by its conformation, molecular orientation, and its oligomeric state in the lipid bilayer. These 'static' structural properties can be routinely studied by solid state NMR using isotope-labeled peptides. In the highly dynamic environment of a liquid crystalline biomembrane, however, the whole-body fluctuations of a peptide are also of paramount importance, although difficult to address and most often ignored. Yet it turns out that disregarding such motional averaging in calculating the molecular alignment from orientational NMR-constraints may give a misleading, if not false picture of the system. Here, we demonstrate that the reliability of a simplified static or an advanced dynamic data analysis depends critically on the choice of isotope labeling scheme used. Two distinctly different scenarios have to be considered. When the labels are placed on the side chains of a helical peptide (such as a CD(3)- or CF(3)-group attached to the C(alpha)C(beta) bond), their nuclear spin interaction tensors are very sensitive to motional averaging. If this effect is not properly accounted for, the helix tilt angle tends to be severely underestimated. At the same time, the analysis of labels in the side chains allows to extract valuable dynamical information about whole-body fluctuations of the peptide helix in the membrane. On the other hand, the alternative labeling scheme where (15)N-labels are accommodated within the peptide backbone, will yield nearly correct helix tilt angles, irrespective as to whether dynamics are taken into account or not - Role of membrane lipids for the activity of pore forming peptides and proteins, Fuertes G, Giménez D, Esteban-Martín S, García-Sáez AJ, Sánchez O & Salgado J.
Adv Exp Med Biol (2010)
677: 31-55.
Bilayer lipids, far from being passive elements, have multiple roles in polypeptide-dependent pore formation. Lipids participate at all stages of the formation of pores by providing the binding site for proteins and peptides, conditioning their active structure and modulating the molecular reorganization of the membrane complex. Such general functions of lipids superimpose to other particular roles, from electrostatic and curvature effects to more specific actions in cases like cholesterol, sphingolipids or cardiolipin. Pores are natural phenomena in lipid membranes. Driven by membrane fluctuations and packing defects, transient water pores are related to spontaneous lipid flip-flop and non-assisted ion permeation. In the absence ofproteins or peptides, these are rare short living events, with properties dependent on the lipid composition of the membrane. Their frequency increases under conditions of internal membrane disturbance of the lipid packing, like in the presence of membrane-bound proteins or peptides. These latter molecules, in fact, form dynamic supramolecular assemblies together with the lipids and transmembrane pores are one of the possible structures of the complex. Active peptides and proteins can thus be considered inducers or enhancers of pores which increase their probability and lifetime by modifying the thermodynamic membrane balance. This includes destabilizing the membrane lamellar structure, lowering the activation energy for pore formation and stabilizing the open pore structure - Pores formed by Baxα5 relax to a smaller size and keep at equilibrium, Fuertes G, García-Sáez AJ, Esteban-Martín S, Giménez D, Sánchez-Muñoz OL, Schwille P & Salgado J.
Biophys J (2010)
99: 2917-2925.
Pores made by amphipathic cationic peptides (e.g., antimicrobials and fragments of pore-forming proteins) are typically studied by examining the kinetics of vesicle leakage after peptide addition or obtaining structural measurements in reconstituted peptide-lipid systems. In the first case, the pores have been considered transient phenomena that allow the relaxation of the peptide-membrane system. In the second, they correspond to equilibrium structures at minimum free energy. Here we reconcile both approaches by investigating the pore activity of the α5 fragment from the proapoptotic protein Bax (Baxα5) before and after equilibrium of peptide/vesicle complexes. Quenching assays on suspensions of large unilamellar vesicles suggest that in the presence of Baxα5, the vesicles maintain a leaky state for hours under equilibrium conditions. We proved and analyzed stable pores on single giant unilamellar vesicles (GUVs) in detail by monitoring the entrance of dyes added at different times after incubation with the peptide. When the GUVs came in contact with Baxα5, leakage started stochastically, was delayed for various periods of time, and in the majority of cases proceeded rapidly to completion. After hours in the presence of the peptide, the same individual GUVs that refilled completely at first instance maintained a porated state, which could be observed in subsequent leak-in events for serially added dyes. However, these long-term pores were smaller in size than the initial equilibration pores. Stable pores were also detected in GUVs made in the presence of Baxα5. The latter pores can be considered equilibrium states and may correspond to structures measured previously in bilayer stacks. Although pore formation may occur as a kinetic process, equilibrium pores may also be functionally relevant structures, especially in highly regulated systems such as the apoptotic mitochondrial pores induced by Bax - Permeabilization of the outer mitochondrial membrane by Bcl-2 proteins, García-Sáez AJ, Fuertes G, Suckale J & Salgado J.
Adv Exp Med Biol (2010)
677: 91-105.
The proteins of the Bcl-2 family regulate the release of the apoptotic factors from mitochondria during apoptosis, a key event in physiological cell death. Although their molecular mechanisms remain unclear, the Bcl-2 proteins have been proposed to directly control the permeability of the outer mitochondrial membrane by pore formation. Indeed, they share structural features with the pore forming domains of some bacterial toxins and they can give rise to proteolipidic pores in model membranes. The complex level of regulation needed to decide the fate of the cell is achieved by an intricate interaction network between different members of the family. Current models consider multiple parallel equilibria of activation and inhibition that determine whether the permeabilization of the mitochondrial outer membrane is induced or not - Structural insights into the activation mechanism of melibiose permease by sodium binding, Granell M, León X, Leblanc G, Padrós E & Lórenz-Fonfría VA.
Proc Natl Acad Sci U S A (2010)
107: 22078-22083.
The melibiose carrier from Escherichia coli (MelB) couples the accumulation of the disaccharide melibiose to the downhill entry of H(+), Na(+), or Li(+). In this work, substrate-induced FTIR difference spectroscopy was used in combination with fluorescence spectroscopy to quantitatively compare the conformational properties of MelB mutants, implicated previously in sodium binding, with those of a fully functional Cys-less MelB permease. The results first suggest that Asp55 and Asp59 are essential ligands for Na(+) binding. Secondly, though Asp124 is not essential for Na(+) binding, this acidic residue may play a critical role, possibly by its interaction with the bound cation, in the full Na(+)-induced conformational changes required for efficient coupling between the ion- and sugar-binding sites; this residue may also be a sugar ligand. Thirdly, Asp19 does not participate in Na(+) binding but it is a melibiose ligand. The location of these residues in two independent threading models of MelB is consistent with their proposed role - Active fragments from pro- and antiapoptotic BCL-2 proteins have distinct membrane behavior reflecting their functional divergence, Guillemin Y, Lopez J, Giménez D, Fuertes G, Valero JG, Blum L, Gonzalo P, Salgado J, Girard-Egrot A & Aouacheria A.
PLoS One (2010)
5: e9066.
The BCL-2 family of proteins includes pro- and antiapoptotic members acting by controlling the permeabilization of mitochondria. Although the association of these proteins with the outer mitochondrial membrane is crucial for their function, little is known about the characteristics of this interaction. Here, we followed a reductionist approach to clarify to what extent membrane-active regions of homologous BCL-2 family proteins contribute to their functional divergence. Using isolated mitochondria as well as model lipid Langmuir monolayers coupled with Brewster Angle Microscopy, we explored systematically and comparatively the membrane activity and membrane-peptide interactions of fragments derived from the central helical hairpin of BAX, BCL-xL and BID. The results show a connection between the differing abilities of the assayed peptide fragments to contact, insert, destabilize and porate membranes and the activity of their cognate proteins in programmed cell death. BCL-2 family-derived pore-forming helices thus represent structurally analogous, but functionally dissimilar membrane domains - Protein fluctuations as the possible origin of the thermal activation of rod photoreceptors in the dark, Lórenz-Fonfría VA, Furutani Y, Ota T, Ido K & Kandori H.
J Am Chem Soc (2010)
132: 5693-5703.
Efficient retinal photoisomerization, signal transduction, and amplification contribute to single-photon electrical responses in vertebrates visual cells. However, spontaneous discrete electrical signals arising in the dark, with identical intensity and time profiles as those generated by genuine single photons (dark events), limit the potential capability of the rod visual system to discern single photons from thermal noise. It is accepted that the light and the thermal activation of the rod photoreceptor rhodopsin (Rho) triggers the light and the dark events, respectively. However the activation barrier for the dark events (80-110 kJ/mol) appears to be only half of the barrier for light-dependent activation of Rho (> or =180 kJ/mol). On the basis of these observations, it has been postulated that both processes should follow different pathways, but the molecular mechanism for the thermal activation process still remains an open question and subject of debate. Here, performing infrared difference spectroscopy measurements, we found that the -OH group of Thr118 from bovine Rho exhibits a slow but measurable hydrogen/deuterium exchange (HDX) under native conditions. Given the location of Thr118 in the X-ray structures, isolated from the aqueous phase and in steric contact with the buried retinal chromophore, we assume that a protein structural fluctuation must drive the retinal binding pocket (RBP) transiently open. We characterized the kinetics (rate and activation enthalpy) and thermodynamics (equilibrium constant and enthalpy) of this fluctuation from the global analysis of the HDX of Thr118-OH as a function of the temperature and pH. In parallel, using HPLC chromatography, we determined the kinetics of the thermal isomerization of the protonated 11-cis retinal in solution, as a model for retinal thermal isomerization in an open RBP. Finally, we propose a quantitative two-step model in which the dark activation of Rho is triggered by thermal isomerization of the retinal in a transiently opened RBP, which accurately reproduced both the experimental activation barrier and the rate of the dark events. We conclude that the absolute sensitivity threshold of our visual system is limited by structural fluctuations of the chromophore binding pocket rather than in the chromophore itself - Time-resolved photoluminescence spectra, lifetime distributions and decay-associated spectra of lanthanide's exchanged microporous-mesoporous materials, Tiseanu C & Lórenz-Fonfria VA.
J Nanosci Nanotechnol (2010)
10: 2803-2810.
Time-resolved luminescence spectra of europium in parent and silylated microporous-mesoporous materials were analyzed by using the maximum entropy method and global multi-exponential fitting, providing lifetime distributions and decay-associated spectra. Silylation was used as a method for the hydrophobization of the materials surface in order to inhibit the moisture intrusion at the lanthanide's sites. Due to the well-known sensitivity of the europium photoluminescence properties to the local environment, our approach can simplify the description of the complex structure-photoluminescence relationships in terms of the individual europium species that contribute to the total emission of the system - Polymer-microporous host interactions probed by photoluminescence spectroscopy, Tiseanu C, Parvulescu VI, Cojocaru B, Lorenz-Fonfria VA, Kumke M, Gessner A & Enculescu I.
Phys Chem Chem Phys (2010)
12: 3031-3037.
Zeolites NaY and ZSM-5 were used as hosts for styrene polymerization after ion-exchange with europium ions. The parent and hybrid, polystyrene coated Eu-NaY (Eu-NaY/PS) and Eu-ZSM-5 (Eu-ZSM-5/PS) zeolites were investigated by using thermal analysis, SEM, PXRD, FT-IR, DR-UV/Vis, steady state and time-resolved photoluminescence spectroscopy. FT-IR spectra evidenced for the interaction between the zeolitic hosts and polystyrene while the PXRD spectra supported for the presence of the polymer inside the channels/pores of Eu-NaY/PS and Eu-ZSM-5/PS materials. The optical properties of Eu-NaY/PS and Eu-ZSM-5/PS were significantly changed relative to those of the parent zeolites, giving further evidence for the presence of polymer inside zeolites. An interesting case is presented by NaY zeolite: following styrene polymerization, the polymer interacted selectively with one of the two main species co-existing inside zeolite while for ZSM-5 a similar effect was not observed - Orientational landscapes of peptides in membranes: prediction of (2)H NMR couplings in a dynamic context, Esteban-Martín S, Giménez D, Fuertes G & Salgado J.
Biochemistry (2009)
48: 11441-11448.
Unlike soluble proteins, membrane polypeptides face an anisotropic milieu. This imposes restraints on their orientation and provides a reference that makes structure prediction tractable by minimalistic thermodynamic models. Here we use this framework to build orientational distributions of monomeric membrane-bound peptides and to predict their expected solid-state (2)H NMR quadrupolar couplings when labeled at specific side chain positions. Using a complete rigid-body sampling of configurations relative to an implicit lipid membrane, peptide free energy landscapes are calculated. This allows us to obtain probability distributions of the peptide tilt, azimuthal rotation, and depth of membrane insertion. The orientational distributions are broad and originate from an interplay among the three relevant rigid-body degrees of freedom, which allows population of multiple states in shallow free energy minima. Remarkably, only when the orientational distributions are taken into account do we obtain a close correlation between predicted (2)H NMR splittings and values measured in experiments. Such a good correlation is not seen with splittings calculated from single configurations, being either the averaged or the lowest free energy state, showing there are distributions, rather than single structures, that best define the peptide-membrane systems. Moreover, we propose that these distributions contribute to the understanding of the rigid-body dynamics of the system - Stability of asymmetric lipid bilayers assessed by molecular dynamics simulations, Esteban-Martín S, Risselada HJ, Salgado J & Marrink SJ.
J Am Chem Soc (2009)
131: 15194-15202.
The asymmetric insertion of amphiphiles into biological membranes compromises the balance between the inner and outer monolayers. As a result, area expansion of the receiving leaflet and curvature strain may lead to membrane permeation, shape changes, or membrane fusion events. We have conducted both atomistic and coarse-grained molecular dynamics simulations of dipalmitoyl-phosphatidylcholine (DPPC) bilayers to study the effect of an asymmetric distribution of lipids between the two monolayers on membrane stability. Highly asymmetric lipid bilayers were found to be surprisingly stable within the submicrosecond time span of the simulations. Even the limiting case of a monolayer immersed in water ruptured spontaneously only after at least 20 ns simulation. A thermal shock could destabilize these kinetically trapped states. We also studied mixed systems composed of DPPC and short tail diC(8)PC lipids, showing that the presence of the cone-shaped short tail lipid facilitates the release of tension in the asymmetric systems via formation of a transmembrane pore. Thus, asymmetric area expansion and curvature stress cooperate to yield bilayer disruption. It appears that, although asymmetric area expansion destabilizes the bilayer structure, the activation energy for transmonolayer lipid re-equilibration is increased. Such a large kinetic barrier can be reduced by lipids with positive spontaneous curvature. These effects are important at the onset of bilayer destabilization phenomena, such as lipid pore formation and membrane fusion, and should be considered for the mechanism of induction of such processes by peptides and proteins - Influence of whole-body dynamics on 15N PISEMA NMR spectra of membrane proteins: a theoretical analysis, Esteban-Martín S, Strandberg E, Fuertes G, Ulrich AS & Salgado J.
Biophys J (2009)
96: 3233-3241.
Membrane proteins and peptides exhibit a preferred orientation in the lipid bilayer while fluctuating in an anisotropic manner. Both the orientation and the dynamics have direct functional implications, but motions are usually not accessible, and structural descriptions are generally static. Using simulated data, we analyze systematically the impact of whole-body motions on the peptide orientations calculated from two-dimensional polarization inversion spin exchange at the magic angle (PISEMA) NMR. Fluctuations are found to have a significant effect on the observed spectra. Nevertheless, wheel-like patterns are still preserved, and it is possible to determine the average peptide tilt and azimuthal rotation angles using simple static models for the spectral fitting. For helical peptides undergoing large-amplitude fluctuations, as in the case of transmembrane monomers, improved fits can be achieved using an explicit dynamics model that includes Gaussian distributions of the orientational parameters. This method allows extracting the amplitudes of fluctuations of the tilt and azimuthal rotation angles. The analysis is further demonstrated by generating first a virtual PISEMA spectrum from a molecular dynamics trajectory of the model peptide, WLP23, in a lipid membrane. That way, the dynamics of the system from which the input spectrum originates is completely known at atomic detail and can thus be directly compared with the dynamic output obtained from the fit. We find that fitting our dynamics model to the polar index slant angles wheel gives an accurate description of the amplitude of underlying motions, together with the average peptide orientation - Spectroscopic and kinetic evidence on how bacteriorhodopsin accomplishes vectorial proton transport under functional conditions, Lórenz-Fonfría VA & Kandori H.
J Am Chem Soc (2009)
131: 5891-5901.
Vectorial transport by pumps requires a switch in the accessibility (or affinity) of the ion binding site from the extracelullar to the cytoplamic side, or vice versa. In the proton-pump bacteriorhodopsin (bR) the nature of this switch mechanism is still controversial, although it is expected to occur during the transition between two M substates. Here, we characterized this transition by time-resolved Fourier transform infrared (FT-IR) spectroscopy under functional conditions, using a novel approach for the analysis of kinetic data: the regularized inversion of an eigenvalue problem. The use of IR spectroscopy allowed the simultaneous evaluation of the involvement of the protein backbone, retinal, amino acid side chains, and internal water molecules in the switch mechanism. We provide solid evidence that the switch is not associated with protein backbone conformational changes. On the other hand, changes in the retinal conformation (or in the orientation of the Schiff Base (SB) during the switch) are reasonably although not completely discarded. We found that the proton release group (PRG), a delocalized proton characterized by a broad continuum band in the infrared, deprotonates in the transition between two M substates. Vectorial proton transport is most likely guaranteed by the coupled proton affinity changes resulting from the PRG deprotonation, favoring an affinity-based over an accessibility-based switch mechanism - In-plane and out-of-plane infrared difference spectroscopy unravels tilting of helices and structural changes in a membrane protein upon substrate binding, Lórenz-Fonfría VA, Granell M, León X, Leblanc G & Padrós E.
J Am Chem Soc (2009)
131: 15094-15095.
Attenuated total reflection infrared (ATR-IR) difference spectroscopy stands out because of its ability to provide information on the interaction of substrates with membrane proteins in their native lipid bilayer environment. We show how the study and interpretation of the structural changes in membrane proteins upon substrate binding is simplified by obtaining ATR-IR difference spectra with polarized light and then computing the difference spectra in the z and x,y directions, where structural and orientation changes give specific difference absorbance patterns. In combination with a maximum-entropy band-narrowing method and some simple spectroscopic rules, the present approach allows us to unambiguously identify changes in the tilt of some helices in the secondary transporter melibiose permease following melibiose binding in the presence of sodium, suggesting the formation of an occluded state during the transport mechanism of the substrates - The role and selection of the filter function in Fourier self-deconvolution revisited, Lórenz-Fonfría VA & Padrós E.
Appl Spectrosc (2009)
63: 791-799.
Overlapped bands often appear in applications of infrared spectroscopy, for instance in the analysis of the amide I band of proteins. Fourier self-deconvolution (FSD) is a popular band-narrowing mathematical method, allowing for the resolution of overlapped bands. The filter function used in FSD plays a significant role in the factor by which the deconvolved bands are actually narrowed (the effective narrowing), as well as in the final signal-to-noise degradation induced by FSD. Moreover, the filter function determines, to a good extent, the band-shape of the deconvolved bands. For instance, the intensity of the harmful side-lobule oscillations that appear in over-deconvolution depends importantly on the filter function used. In the present paper we characterized the resulting band shape, effective narrowing, and signal-to-noise degradation in infra-, self-, and over-deconvolution conditions for several filter functions: Triangle, Bessel, Hanning, Gaussian, Sinc2, and Triangle2. We also introduced and characterized new filters based on the modification of the Blackmann filter. Our conclusion is that the Bessel filter (in infra-, self-, and mild over-deconvolution), the newly introduced BL3 filter (in self- and mild/moderate over-deconvolution), and the Gaussian filter (in moderate/strong over-deconvolution) are the most suitable filter functions to be used in FSD - Orientation and dynamics of peptides in membranes calculated from 2H-NMR data, Strandberg E, Esteban-Martín S, Salgado J & Ulrich AS.
Biophys J (2009)
96: 3223-3232.
Solid-state (2)H-NMR is routinely used to determine the alignment of membrane-bound peptides. Here we demonstrate that it can also provide a quantitative measure of the fluctuations around the distinct molecular axes. Using several dynamic models with increasing complexity, we reanalyzed published (2)H-NMR data on two representative alpha-helical peptides: 1), the amphiphilic antimicrobial peptide PGLa, which permeabilizes membranes by going from a monomeric surface-bound to a dimeric tilted state and finally inserting as an oligomeric pore; and 2), the hydrophobic WALP23, which is a typical transmembrane segment, although previous analysis had yielded helix tilt angles much smaller than expected from hydrophobic mismatch and molecular dynamics simulations. Their (2)H-NMR data were deconvoluted in terms of the two main helix orientation angles (representing the time-averaged peptide tilt and azimuthal rotation), as well as the amplitudes of fluctuation about the corresponding molecular axes (providing the dynamic picture). The mobility of PGLa is found to be moderate and to correlate well with the respective oligomeric states. WALP23 fluctuates more vigorously, now in better agreement with the molecular dynamics simulations and mismatch predictions. The analysis demonstrates that when (2)H-NMR data are fitted to extract peptide orientation angles, an explicit representation of the peptide rigid-body angular fluctuations should be included
2007-08
- Solid State NMR Structure Analysis of the Antimicrobial Peptide Gramicidin S in Lipid Membranes: Concentration-Dependent Re-alignment and Self-Assembly as a β-Barrel, Afonin S, Dürr UHN, Wadhwani P, Salgado J & Ulrich AS.
Top Curr Chem (2008)
273: 139-154.
Antimicrobial peptides can kill bacteria by permeabilizing their cell membrane, as these amphiphilicmolecules interact favourably with lipid bilayers. This mechanism of action is attributed eitherto the formation of a peptide 'carpet' on the membrane surface, or to a transmembranepore. However, the structure of such a pore has not yet been resolved under relevant conditions.Gramicidin S is a symmetrical cyclic β-sheet decapeptide, which has been previouslyshown by solid state NMR to lie flat on the membrane surface at low peptide:lipid ratios (≤ 1:80).Using highly sensitive (19)F-NMR, supported by (15)N-labelling,we found that gramicidin S can flip into an upright transmembrane alignment at high peptide:lipidratios (≥ 1:40). Orientational NMR constraints suggest that the peptide may self-assembleas an oligomeric β-barrel pore, which is stabilized by intermolecular hydrogen bonds. Comparisonof different model membranes shows that the observed re-alignment is favoured in thin bilayers withshort-chain lipids, especially near the chain melting temperature, whereas long-chain lipids suppresspore formation. Based on the oligomeric structural model and the conditions of pore formation, guidelinesmay now be derived for rationally designing peptide analogues as antibiotics with improved selectivityand reduced side effects - FTIR spectroscopy of secondary-structure reorientation of melibiose permease modulated by substrate binding, Dave N, Lórenz-Fonfría VA, Leblanc G & Padrós E.
Biophys J (2008)
94: 3659-3670.
Analysis of infrared polarized absorbance spectra and linear dichroism spectra of reconstituted melibiose permease from Escherichia coli shows that the oriented structures correspond mainly to tilted transmembrane alpha-helices, forming an average angle of approximately 26 degrees with the membrane normal in substrate-free medium. Examination of the deconvoluted linear dichroism spectra in H(2)O and D(2)O makes apparent two populations of alpha-helices differing by their tilt angle (helix types I and II). Moreover, the average helical tilt angle significantly varies upon substrate binding: it is increased upon Na(+) binding, whereas it decreases upon subsequent melibiose binding in the presence of Na(+). In contrast, melibiose binding in the presence of H(+) causes virtually no change in the average tilt angle. The data also suggest that the two helix populations change their tilting and H/D exchange level in different ways depending on the bound substrate(s). Notably, cation binding essentially influences type I helices, whereas melibiose binding modifies the tilting of both helix populations - Method for the estimation of the mean lorentzian bandwidth in spectra composed of an unknown number of highly overlapped bands, Lórenz-Fonfría VA & Padrós E.
Appl Spectrosc (2008)
62: 689-700.
We introduce a method for the estimation of the mean Lorentzian bandwidth of the component bands in a spectrum. The method is computationally simple, using only the module of the Fourier transform of the spectrum, and its first derivative. Moreover, the presented method does not require knowledge of the number of bands in the spectrum, their band positions, or their band areas. Furthermore, it works on spectra containing Lorentzian bands, as well as Gaussian and Voigtian bands. Therefore, the introduced method seems especially well suited for obtaining a representative Lorentzian width for highly overlapped bands, independent of their number and Lorentzian/Gaussian character. We describe how different experimental limitations (spectral truncation, offset error, presence of noise, etc.) may affect the performance of the method, and when required we propose effective alternatives to minimize their effects. Finally, we show the application of the method to an experimental spectrum: the amide I band of a dry film of the solubilized ADP/ATP carrier. The estimation of the mean Lorentzian width can allow, for instance, for a more objective selection of the deconvolution width in Fourier self-deconvolution, allowing for a more objective and reliable analysis of the amide I band of proteins. The mean Lorentzian width can also be useful to obtain an estimation of the homogenous broadening and vibrational relaxation of the amide I vibration of proteins, without requiring complex pump-probe experiments - Active internal waters in the bacteriorhodopsin photocycle. A comparative study of the L and M intermediates at room and cryogenic temperatures by infrared spectroscopy, Lórenz-Fonfría VA, Furutani Y & Kandori H.
Biochemistry (2008)
47: 4071-4081.
We present time-resolved room-temperature infrared difference spectra for the bacteriorhodopsin (bR) photocycle at 8 cm (-1) spectral and 5 micros temporal resolution, from 4000 to 800 cm (-1). An in situ hydration method allowed for a controlled and stable sample hydration (92% relative humidity), largely improving the quality of the data without affecting the functionality of bR. Experiments in both H 2 (16)O and H 2 (18)O were conducted to assign bands to internal water molecules. Room-temperature difference spectra of the L and M intermediates minus the bR ground state (L-BR and M-BR, respectively) were comprehensively compared with their low-temperature counterparts. The room-temperature M-BR spectrum was almost identical to that obtained at 230 K, except for a continuum band. The continuum band contains water vibrations from this spectral comparison between H 2 (16)O and H 2 (18)O, and no continuum band at 230 K suggests that the protein/solvent dynamics are insufficient for deprotonation of the water cluster. On the other hand, an intense positive broadband in the low-temperature L-BR spectrum (170 K) assigned to the formation of a water cavity in the cytoplasmic domain is absent at room temperature. This water cavity, proposed to be an essential feature for the formation of L, seems now to be a low-temperature artifact caused by restricted protein dynamics at 170 K. The observed differences between low- and room-temperature FTIR spectra are further discussed in light of previously reported dynamic transitions in bR. Finally, we show that the kinetics of the transient heat relaxation of bR after photoexcitation proceeds as a thermal diffusion process, uncorrelated with the photocycle itself - Electronic Properties in a Five-Coordinate Azido Complex of Nonplanar Iron(III) Porphyrin: Revisiting to Quantum Mechanical Spin Admixing, Neya S, Takahashi A, Ode H, Hoshino T, Ikezaki A, Ohgo Y, Takahashi M, Furutani Y, Lórenz-Fonfría VA, Kandori H, Hiramatsu H, Kitagawa T, Teraoka J, Funasaki N & Nakamura M.
Bull Chem Soc Jpn (2008)
81: 136-141.
The iron(III) azido complex of 5,10,15,20-tetraisopropylporphyrin was characterized with NMR, EPR, Mössbauer, and magnetic susceptibility. These physical methods indicate mixing of the high (S=5⁄2) and intermediate (S=3⁄2) spin-states of the iron atom. The results were interpreted in terms of the core contraction after nonplanar deformation of porphyrin ring by the bulky isopropyl substituents. In the IR spectrum of this complex, there are two signals at 2062 and 2048 cm−1 due to the antisymmetric vibration of the coordinated azido ligand. The split IR bands demonstrate that the two spin isomers are present, and that the S=5⁄2 and 3/2 transition occurs sufficiently slow on the IR timescale. This is in remarkable contrast with the homogeneous spin-mixing model proposed for the S=5⁄2 and 3/2 system. The present observations further suggests that the three S=5⁄2, 3/2, and 1/2 states in iron(III) porphyrin commonly mix through thermal spin equilibrium. - Design of a bivalent peptide with two independent elements of secondary structure able to fold autonomously, Pantoja-Uceda D, Pastor MT, Salgado J, Pineda-Lucena A & Pérez-Payá E.
J Pept Sci (2008)
14: 845-854.
This article describes a strategy to develop, starting from a de novo design, bivalent peptides containing two different (alpha-helix and beta-hairpin) and independent secondary-structure elements. The design was based on the use of conformationally restricted peptide libraries. Structural characterization by NMR revealed that the peptides were stable and did not show any long-range NOE interactions between the N-terminal beta-hairpin and the C-terminal alpha-helix. These results suggest that the two elements of secondary structure are stable and well folded - Influence of proline on the thermostability of the active site and membrane arrangement of transmembrane proteins, Perálvarez-Marín A, Lórenz-Fonfría VA, Simón-Vázquez R, Gomariz M, Meseguer I, Querol E & Padrós E.
Biophys J (2008)
95: 4384-4395.
Proline residues play a fundamental and subtle role in the dynamics, structure, and function in many membrane proteins. Temperature derivative spectroscopy and differential scanning calorimetry have been used to determine the effect of proline substitution in the structural stability of the active site and transmembrane arrangement of bacteriorhodopsin. We have analyzed the Pro-to-Ala mutation for the helix-embedded prolines Pro50, Pro91, and Pro186 in the native membrane environment. This information has been complemented with the analysis of the respective crystallographic structures by the FoldX force field. Differential scanning calorimetry allowed us to determine distorted membrane arrangement for P50A and P186A. The protein stability was severely affected for P186A and P91A. In the case of Pro91, a single point mutation is capable of strongly slowing down the conformational diffusion along the denaturation coordinate, becoming a barrier-free downhill process above 371 K. Temperature derivative spectroscopy, applied for first time to study thermal stability of proteins, has been used to monitor the stability of the active site of bacteriorhodopsin. The mutation of Pro91 and Pro186 showed the most striking effects on the retinal binding pocket. These residues are the Pro in closer contact to the active site (activation energies for retinal release of 60.1 and 76.8 kcal/mol, respectively, compared to 115.8 kcal/mol for WT). FoldX analysis of the protein crystal structures indicates that the Pro-to-Ala mutations have both local and long-range effects on the structural stability of residues involved in the architecture of the protein and the active site and in the proton pumping function. Thus, this study provides a complete overview of the substitution effect of helix-embedded prolines in the thermodynamic and dynamic stability of a membrane protein, also related to its structure and function - Photoluminescence study of terbium-exchanged ultrastable Y zeolites: Number of species, photoluminescence decays, and decay-associated spectra, Tiseanu C, Lórenz-Fonfría VA, Parvulescu VI, Gessner A & Kumke MU.
J Appl Phys (2008)
104: 033530.
Terbium-exchanged ultrastable Y (USY) zeolites were investigated by using time-resolved photoluminescence spectroscopy techniques and methods. To determine the distribution of terbium species in USY zeolites together with their photoluminescence properties, several analysis methods for the time-resolved luminescence spectra were used such as the area normalization of time-resolved photoluminescence spectra, singular value decomposition, global nonlinear least squares, and the maximum entropy. Except for a questionable long lifetime, small contribution of a terbium species with lifetime of 1.9–2.1 ms, all the experimental and analysis results converged to a two terbium species distribution with lifetimes varying between 410–440 and 1000–1100 μs. The effects of the silylation of terbium-exchanged USY zeolites with phenyl-, vinyl-, and hexadecyltrimethoxysilanes on the lanthanide’s photoluminescence properties were also described. - Comparative luminescence study of terbium-exchanged zeolites silylated with alkoxysilanes, Tiseanu C, Lórenz-Fonfría VA, Gessner A, Kumke MU & Gagea B.
J Mater Sci : Mater Electron (2008)
20: 312-316.
Terbium-exchanged ZSM-5, MOR and (H)BEA zeolites were silylated with phenyl-, vinyl- and hexadecyl trimethoxysilanes via a post-synthesis grafting. All samples were investigated by means of PXRD, FT-IR, TGA, physical adsorption, DR-UV-Vis and time-resolved photoluminescence spectroscopy. From the comparison of the photoluminescence decays of terbium-exchanged in parent (non-silylated) and silylated zeolites, it resulted that the silylation efficiency of the various alkoxysilanes is determined by the type of zeolite and follows the sequences: phenyl > vinyl > hexadecyl > parent for ZSM-5, hexadecyl ≈ phenyl ≈ vinyl > parent for MOR and hexadecyl > phenyl ≈ vinyl >≈ parent for BEA zeolites, respectively. - The dynamic orientation of membrane-bound peptides: bridging simulations and experiments, Esteban-Martín S & Salgado J.
Biophys J (2007)
93: 4278-4288.
The structural organization in a peptide/membrane supramolecular complex is best described by knowledge of the peptide orientation plus its time-dependent and spatial fluctuations. The static orientation, defined by the peptide tilt and a rotation about its molecular axis, is accessible through a number of spectroscopic methods. However, peptide dynamics, although relevant to understand the functionality of these systems, remains largely unexplored. Here, we describe the orientation and dynamics of Trp-flanked and Lys-flanked hydrophobic peptides in a lipid bilayer from molecular dynamics simulations. A novel view is revealed, where collective nontrivial distributions of time-evolving and ensemble peptide orientations closely represent the systems as studied experimentally. Such global distributions are broad and unveil the existence of orientational states, which depend on the anchoring mode of interfacial residues. We show that this dynamics modulates (2)H quadrupolar splittings and introduces ambiguity in the analysis of NMR data. These findings demonstrate that structural descriptions of peptide/membrane complexes are incomplete, and in cases even imprecise, without knowledge of dynamics - Self-assembling of peptide/membrane complexes by atomistic molecular dynamics simulations, Esteban-Martín S & Salgado J.
Biophys J (2007)
92: 903-912.
Model biological membranes consisting of peptide/lipid-bilayer complexes can nowadays be studied by classical molecular dynamics (MD) simulations at atomic detail. In most cases, the simulation starts with an assumed state of a peptide in a preformed bilayer, from which equilibrium configurations are difficult to obtain due to a relatively slow molecular diffusion. As an alternative, we propose an extension of reported work on the self-organization of unordered lipids into bilayers, consisting of including a peptide molecule in the initial random configuration to obtain a membrane-bound peptide simultaneous to the formation of the lipid bilayer. This strategy takes advantage of the fast reorganization of lipids, among themselves and around the peptide, in an aqueous environment. Model peptides of different hydrophobicity, CH3-CO-W2L18W2-NH2 (WL22) and CH3-CO-W2A18W2-NH2 (WA22), in dipalmitoyl-phosphatidylcholine (DPPC) are used as test cases. In the equilibrium states of the peptide/membrane complexes, achieved in time ranges of 50-100 ns, the two peptides behave as expected from experimental and theoretical studies. The strongly hydrophobic WL22 is inserted in a transmembrane configuration and the marginally apolar, alanine-based WA22 is found in two alternative states: transmembrane inserted or parallel to the membrane plane, embedded close to the bilayer interface, with similar stability. This shows that the spontaneous assembly of peptides and lipids is an unbiased and reliable strategy to produce and study models of equilibrated peptide/lipid complexes of unknown membrane-binding mode and topology - Pore formation by a Bax-derived peptide: effect on the line tension of the membrane probed by AFM, García-Sáez AJ, Chiantia S, Salgado J & Schwille P.
Biophys J (2007)
93: 103-112.
Bax is a critical regulator of physiological cell death that increases the permeability of the outer mitochondrial membrane and facilitates the release of the so-called apoptotic factors during apoptosis. The molecular mechanism of action is unknown, but it probably involves the formation of partially lipidic pores induced by Bax. To investigate the interaction of Bax with lipid membranes and the physical changes underlying the formation of Bax pores, we used an active peptide derived from helix 5 of this protein (Bax-alpha5) that is able to induce Bax-like pores in lipid bilayers. We report the decrease of line tension due to peptide binding both at the domain interface in phase-separated lipid bilayers and at the pore edge in atomic force microscopy film-rupture experiments. Such a decrease in line tension may be a general strategy of pore-forming peptides and proteins, as it affects the energetics of the pore and stabilizes the open state - Bayesian maximum entropy (two-dimensional) lifetime distribution reconstruction from time-resolved spectroscopic data, Lórenz-Fonfría VA & Kandori H.
Appl Spectrosc (2007)
61: 428-443.
Time-resolved spectroscopy is often used to monitor the relaxation processes (or reactions) of physical, chemical, and biochemical systems after some fast physical or chemical perturbation. Time-resolved spectra contain information about the relaxation kinetics, in the form of macroscopic time constants of decay and their decay associated spectra. In the present paper we show how the Bayesian maximum entropy inversion of the Laplace transform (MaxEnt-iLT) can provide a lifetime distribution without sign-restrictions (or two-dimensional (2D)-lifetime distribution), representing the most probable inference given the data. From the reconstructed (2D) lifetime distribution it is possible to obtain the number of exponentials decays, macroscopic rate constants, and exponential amplitudes (or their decay associated spectra) present in the data. More importantly, the obtained (2D) lifetime distribution is obtained free from pre-conditioned ideas about the number of exponential decays present in the data. In contrast to the standard regularized maximum entropy method, the Bayesian MaxEnt approach automatically estimates the regularization parameter, providing an unsupervised and more objective analysis. We also show that the regularization parameter can be automatically determined by the L-curve and generalized cross-validation methods, providing (2D) lifetime reconstructions relatively close to the Bayesian best inference. Finally, we propose the use of MaxEnt-iLT for a more objective discrimination between data-supported and data-unsupported quantitative kinetic models, which takes both the data and the analysis limitations into account. All these aspects are illustrated with realistic time-resolved Fourier transform infrared (FT-IR) synthetic spectra of the bacteriorhodopsin photocycle - Practical aspects of the maximum entropy inversion of the laplace transform for the quantitative analysis of multi-exponential data, Lórenz-Fonfría VA & Kandori H.
Appl Spectrosc (2007)
61: 74-84.
The number, position, area, and width of the bands in a lifetime distribution give the number of exponentials present in time-resolved data and their time constants, amplitudes, and heterogeneities. The maximum entropy inversion of the Laplace transform (MaxEnt-iLT) provides a lifetime distribution from time-resolved data, which is very helpful in the analysis of the relaxation of complex systems. In some applications both positive and negative values for the lifetime distribution amplitudes are physical, but most studies to date have focused on positive-constrained solutions. In this work, we first discuss optimal conditions to obtain a sign-unrestricted maximum entropy lifetime distribution, i.e., the selection of the entropy function and the regularization value. For the selection of the regularization value we compared four methods: the chi2 criterion and Bayesian inference (already used in sign-restricted MaxEnt-iLT), and the L-curve and the generalized cross-validation methods (not yet used in MaxEnt-iLT to our knowledge). Except for the frequently used chi2 criterion, these methods recommended similar regularization values, providing close to optimum solutions. However, even when an optimal entropy function and regularization value are used, a MaxEnt lifetime distribution will contain noise-induced errors, as well as systematic distortions induced by the entropy maximization (regularization-induced errors). We introduce the concept of the apparent resolution function in MaxEnt, which allows both the noise and regularization-induced errors to be estimated. We show the capability of this newly introduced concept in both synthetic and experimental time-resolved Fourier transform infrared (FT-IR) data from the bacteriorhodopsin photocycle - Inter-helical hydrogen bonds are essential elements for intra-protein signal transduction: the role of Asp115 in bacteriorhodopsin transport function, Perálvarez-Marín A, Lórenz-Fonfría VA, Bourdelande J-L, Querol E, Kandori H & Padrós E.
J Mol Biol (2007)
368: 666-676.
The behavior of the D115A mutant was analyzed by time-resolved UV-Vis and Fourier transformed infrared (FTIR) spectroscopies, aiming to clarify the role of Asp115 in the intra-protein signal transductions occurring during the bacteriorhodopsin photocycle. UV-Vis data on the D115A mutant show severely desynchronized photocycle kinetics. FTIR data show a poor transmission of the retinal isomerization to the chromoprotein, evidenced by strongly attenuated helical changes (amide I), the remarkable absence of environment alterations and protonation/deprotonation events related to Asp96 and direct Schiff base (SB) protonation form the bulk. This argues for the interactions of Asp115 with Leu87 (via water molecule) and Thr90 as key elements for the effective and vectorial proton path between Asp96 and the SB, in the cytoplasmic half of bacteriorhodopsin. The results strongly suggest the presence of a regulation motif enclosed in helices C and D (Thr90-Pro91/Asp115) which drives properly the dynamics of helix C through a set of interactions. It also supports the idea that intra-helical hydrogen bonding clusters in the buried regions of transmembrane proteins can be potential elements in intra-protein signal transduction - Vanillin cell sensor, Rodrigo G, Carrera J, Gimenez D, Fernandez-de-Cordoba P, Salgado J, Montagud A, Urchueguia J, Aroca MC, Mata C, Ferrando A, Navarrete C, Tortosa P, Baguena M, Jaramillo A, Fuertes G, Edo C, Medrano JV, Navarro E & Aparici A.
IET Synth Biol (2007)
1: 74-78.
Our project for iGEM 2006 consisted of designing a cellular vanillin biosensor. We used an EnvZ–E. coli strain as a chassis, and constructed two different devices: a sensor and an actuator, assembled using OmpR-P as a standardised mediator. The sensor device contained a computationally designed vanillin receptor and a synthetic two-component signal transduction protein (Trz). The receptor protein was based on a ribose-binding protein as scaffold. The Trz was built by fusion of the periplasmic and transmembrane domains of a Trg protein with an EnvZ kinase domain. When the receptor complex binds Trg, an allosteric motion is propagated to the cytoplasmic EnvZ kinase domain, resulting in autophosphorylation and subsequent phosphate transfer to the OmpR transcription factor, which finally induces transcription of the ompC promoter. As actuator, we used a synthetic transcriptional circuit, which implements an OmpR-P band detector having GFP and RFP as an output. We designed this circuit using a synthetic promoter working as an AND gate, which is synergistically activated by cI and CRP. Our constructed Trg-EnvZ fusion and AND promoter will be very useful to future synthetic biology projects - Investigation of the Hydrophobization Efficiency of Terbium-Exchanged BEA Zeolites by Means of FT-IR, TGA, Physical Adsorption, and Time-Resolved Photoluminescence, Tiseanu C, Gagea B, Parvulescu VI, Lórenz-Fonfría V, Gessner A & Kumke MU.
Langmuir (2007)
23: 6781-6787.
Terbium-exchanged BEA zeolites were hydrophobized with phenyl-, vinyl-, and hexadecyltrimethoxysilanes by means of postsynthesis grafting. These materials were investigated using XRD, FT-IR, TGA, physical adsorption, and photoluminescence. Different methods for the analysis of the non-exponential decay of terbium photoluminescence in BEA zeolites were used ranging from discrete exponential to more complex approaches based on maximum entropy and global analysis. Two groups of decay times varying between 480 and 580 μs and 1−1.3 ms were assigned to the lifetimes of terbium exposed to water (unprotected) and protected by the organic groups, respectively. Our results showed that the preservation of terbium PL properties against detrimental effects of moisture adsorption could be ordered in the following sequence: hexadecyl > phenyl ≈ vinyl. The photoluminescence results were in good agreement with the FT-IR, TGA, and physical adsorption data.
2005-06
- Peptides corresponding to helices 5 and 6 of Bax can independently form large lipid pores, García-Sáez AJ, Coraiola M, Dalla Serra M, Mingarro I, Müller P & Salgado J.
FEBS J (2006)
273: 971-981.
Proteins of the B-cell lymphoma protein 2 (Bcl2) family are key regulators of the apoptotic cascade, controlling the release of apoptotic factors from the mitochondrial intermembrane space. A helical hairpin found in the core of water-soluble folds of these proteins has been reported to be the pore-forming domain. Here we show that peptides including any of the two alpha-helix fragments of the hairpin of Bcl2 associated protein X (Bax) can independently induce release of large labelled dextrans from synthetic lipid vesicles. The permeability promoted by these peptides is influenced by intrinsic monolayer curvature and accompanied by fast transbilayer redistribution of lipids, supporting a toroidal pore mechanism as in the case of the full-length protein. However, compared with the pores made by complete Bax, the pores made by the Bax peptides are smaller and do not need the concerted action of tBid. These data indicate that the sequences of both fragments of the hairpin contain the principal physicochemical requirements for pore formation, showing a parallel between the permeabilization mechanism of a complex regulated protein system, such as Bax, and the much simpler pore-forming antibiotic peptides - Transformation of time-resolved spectra to lifetime-resolved spectra by maximum entropy inversion of the laplace transform, Lórenz-Fonfría VA & Kandori H.
Appl Spectrosc (2006)
60: 407-417.
We present a method for the analysis of time-resolved spectroscopic data following first-order kinetics. The time traces at all the available spectroscopic channels (e.g., wavelength or wavenumber) are inverse Laplace transformed. The transformation is stabilized by the maximum entropy method generalized for solutions without sign-restriction. In this way, time-resolved spectra can be converted to lifetime-resolved spectra, where bands appear at coordinates corresponding to their spectroscopic maxima and time constant of appearance (negative amplitude) or disappearance (positive amplitude). From the lifetime-resolved spectra, the number of exponentially decaying components, their time constants, and their decay-associated spectra are readily available. Moreover, since bands are spread in two dimensions extra band-resolution is possible. We named this method of transforming time-resolved spectra into lifetime-resolved spectra multi-spectroscopic channel maximum entropy inversion of the Laplace transform (M-MaxEnt-iLT). The basis of M-MaxEnt-iLT is presented in detail and its properties and limitations are thoroughly discussed. We also show how the combination of M-MaxEnt-iLT with spectral smoothing or deconvolution can improve the appearance and/or band resolution of the obtained lifetime-resolved spectra - Time-resolved rapid-scan Fourier transform infrared difference spectroscopy on a noncyclic photosystem: rhodopsin photointermediates from Lumi to Meta II, Lüdeke S, Lórenz Fonfría VA, Siebert F & Vogel R.
Biopolymers (2006)
83: 159-169.
The visual pigment rhodopsin has been extensively studied for the kinetics of its photointermediates by various spectroscopic methods. Unlike such archaeal retinal proteins as bacteriorhodopsin, visual rhodopsin does not thermally recover its dark state after photoexcitation, which precludes repeated excitation of a single sample and thereby complicates time-resolved experiments. Kinetic data on the late rhodopsin photointermediates have so far been available mainly from time-resolved ultraviolet (UV)-visible spectroscopy, but not from Fourier transform infrared (FTIR) spectroscopy. The latter has the advantage of being informative of structural changes of both chromophore and protein, but does not allow the highly reproducible, automated sample exchange procedures available to UV-visible spectroscopy. Using rapid-scan FTIR difference spectroscopy, we obtained time-resolved data sets that were analyzed by a maximum entropy inverse Laplace-transform. Covering the time range from 8 ms to 15 s at temperatures of 0 and -7 degrees C, the transitions from the Lumi to the Meta I and from the Meta I to the Meta II photoproduct states could be resolved. In the transition from Meta I to Meta II, our data reveal a partial deprotonation of the retinal Schiff base preceding the conformational change of the receptor protein to Meta II. The technique and the results are discussed in regard to its advantages as well as its limitations - Production and characterisation of recombinant forms of human pulmonary surfactant protein C (SP-C): Structure and surface activity, Lukovic D, Plasencia I, Taberner FJ, Salgado J, Calvete JJ, Pérez-Gil J & Mingarro I.
Biochim Biophys Acta, Biomembr (2006)
1758: 509-518.
Surfactant protein C (SP-C) is an essential component for the surface tension-lowering activity of the pulmonary surfactant system. It contains a valine-rich alpha helix that spans the lipid bilayer, and is one of the most hydrophobic proteins known so far. SP-C is also an essential component of various surfactant preparations of animal origin currently used to treat neonatal respiratory distress syndrome (NRDS) in preterm infants. The limited supply of this material and the risk of transmission of infectious agents and immunological reactions have prompted the development of synthetic SP-C-derived peptides or recombinant humanized SP-C for inclusion in new preparations for therapeutic use. We describe herein the recombinant production in bacterial cultures of SP-C variants containing phenylalanines instead of the palmitoylated cysteines of the native protein, as fusions to the hydrophilic nuclease A (SN) from Staphylococcus aureus. The resulting chimerae were partially purified by affinity chromatography and subsequently subjected to protease digestion. The SP-C forms were recovered from the digestion mixtures by organic extraction and further purified by size exclusion chromatography. The two recombinant SP-C variants so obtained retained more than 50% alpha-helical content and showed surface activity comparable to the native protein, as measured by surface spreading of lipid/protein suspensions and from compression pi-A isotherms of lipid/protein films. Compared to the protein purified from porcine lungs, the recombinant SP-C forms improved movement of phospholipid molecules into the interface (during adsorption), or out from the interfacial film (during compression), suggesting new possibilities to develop improved therapeutic preparations - Peptides derived from apoptotic Bax and Bid reproduce the poration activity of the parent full-length proteins, García-Sáez AJ, Coraiola M, Dalla Serra M, Mingarro I, Menestrina G & Salgado J.
Biophys J (2005)
88: 3976-3990.
Bax and Bid are proapoptotic proteins of the Bcl-2 family that regulate the release of apoptogenic factors from mitochondria. Although they localize constitutively in the cytoplasm, their apoptotic function is exerted at the mitochondrial outer membrane, and is related to their ability to form transbilayer pores. Here we report the poration activity of fragments from these two proteins, containing the first alpha-helix of a colicinlike hydrophobic hairpin (alpha-helix 5 of Bax and alpha-helix 6 of Bid). Both peptides readily bind to synthetic lipid vesicles, where they adopt predominantly alpha-helical structures and induce the release of entrapped calcein. In planar lipid membranes they form ion conducting channels, which in the case of the Bax-derived peptide are characterized by a two-stage pattern, a large conductivity and lipid-charge-dependent ionic selectivity. These features, together with the influence of intrinsic lipid curvature on the poration activity and the existence of two helical stretches of different orientations for the membrane-bound peptide, suggest that it forms mixed lipidic/peptidic pores of toroidal structure. In contrast, the assayed Bid fragment shows a markedly different behavior, characterized by the formation of discrete, steplike channels in planar lipid bilayers, as expected for a peptidic pore lined by a bundle of helices - Substrate-induced conformational changes of melibiose permease from Escherichia coli studied by infrared difference spectroscopy, León X, Lórenz-Fonfría VA, Lemonnier R, Leblanc G & Padrós E.
Biochemistry (2005)
44: 3506-3514.
Fourier transform infrared difference spectroscopy has been used to obtain information about substrate-induced structural changes of the melibiose permease (MelB) from Escherichia coli reconstituted into liposomes. Binding of the cosubstrate Na(+) gives rise to several peaks in the amide I and II regions of the difference spectrum Na(+).MelB minus H(+).MelB, that denote the presence of conformational changes in all types of secondary structures (alpha-helices, beta-sheets, loops). In addition, peaks around 1400 and at 1740-1720 cm(-1) are indicative of changes in protonation/deprotonation or in environment of carboxylic groups. Binding of the cosubstrate Li(+) produces a difference spectrum that is also indicative of conformational changes, but that is at variance as compared to that induced by Na(+) binding. To analyze the following transport steps, the melibiose permease with either H(+), Na(+), or Li(+) bound was incubated with melibiose. The difference spectra obtained by subtracting the spectrum cation.MelB from the respective complex cation.melibiose.MelB were roughly similar among them, but different from those induced by cation binding, and more intense. Therefore, major conformational changes that are induced during melibiose binding/substrate translocation, like those denoted by intense peaks at 1668 and 1645 cm(-)(1), are similar for the three cotransporting cations. Changes in the protonation state and/or in the environment of given carboxylic residues were also induced by melibiose-MelB interaction in the presence of cations - Maximum entropy deconvolution of infrared spectra: use of a novel entropy expression without sign restriction, Lórenz-Fonfría VA & Padrós E.
Appl Spectrosc (2005)
59: 474-486.
Absorbance and difference infrared spectra are often acquired aiming to characterize protein structure and structural changes of proteins upon ligand binding, as well as for many other chemical and biochemical studies. Their analysis requires as a first step the identification of the component bands (number, position, and area) and as a second step their assignment. The first step of the analysis is challenged by the habitually strong band overlap in infrared spectra. Therefore, it is useful to make use of a mathematical method able to narrow the component bands to the extent to eliminate, or at least reduce, the band overlap. Additionally, to be of general applicability this method should permit negative values for the solution. We present a maximum entropy deconvolution approach for the band-narrowing of absorbance and difference spectra showing the required characteristics, which uses the generalized negative Burg-entropy (Itakura-Saito discrepancy) generalized for difference spectra. We present results on synthetic noisy absorbance and difference spectra, as well as on experimental infrared spectra from the membrane protein bacteriorhodopsin - Double-spanning plant viral movement protein integration into the endoplasmic reticulum membrane is signal recognition particle-dependent, translocon-mediated, and concerted, Saurí A, Saksena S, Salgado J, Johnson AE & Mingarro I.
J Biol Chem (2005)
280: 25907-25912.
The current model for cell-to-cell movement of plant viruses holds that transport requires virus-encoded movement proteins that intimately associate with endoplasmic reticulum membranes. We have examined the early stages of the integration into endoplasmic reticulum membranes of a double-spanning viral movement protein using photocross-linking. We have discovered that this process is cotranslational and proceeds in a signal recognition particle-dependent manner. In addition, nascent chain photocross-linking to Sec61alpha and translocating chain-associated membrane protein reveal that viral membrane protein insertion takes place via the translocon, as with most eukaryotic membrane proteins, but that the two transmembrane segments of the viral protein leave the translocon and enter the lipid bilayer together
2003-04
- Membrane-insertion fragments of Bcl-xL, Bax, and Bid, García-Sáez AJ, Mingarro I, Pérez-Payá E & Salgado J.
Biochemistry (2004)
43: 10930-10943.
Apoptosis regulators of the Bcl-2 family associate with intracellular membranes from mitochondria and the endoplasmic reticulum, where they perform their function. The activity of these proteins is related to the release of apoptogenic factors, sequestered in the mitochondria, to the cytoplasm, probably through the formation of ion and/or protein transport channels. Most of these proteins contain a C-terminal putative transmembrane (TM) fragment and a pair of hydrophobic alpha helices (alpha5-alpha6) similar to the membrane insertion fragments of the ion-channel domain of diphtheria toxin and colicins. Here, we report on the membrane-insertion properties of different segments from antiapoptotic Bcl-x(L) and proapoptotic Bax and Bid, that correspond to defined alpha helices in the structure of their soluble forms. According to prediction methods, there are only two putative TM fragments in Bcl-x(L) and Bax (the C-terminal alpha helix and alpha-helix 5) and one in activated tBid (alpha-helix 6). The rest of their sequence, including the second helix of the pore-forming domain, displays only weak hydrophobic peaks, which are below the prediction threshold. Subsequent analysis by glycosylation mapping of single alpha-helix segments in a model chimeric system confirms the above predictions and allows finding an extra TM fragment made of helix alpha1 of Bax. Surprisingly, the amphipathic helices alpha6 of Bcl-x(L) and Bax and alpha7 of Bid do insert in membranes only as part of the alpha5-alpha6 (Bcl-x(L) and Bax) or alpha6-alpha7 (Bid) hairpins but not when assayed individually. This behavior suggests a synergistic insertion and folding of the two helices of the hairpin that could be due to charge complementarity and additional stability provided by turn-inducing residues present at the interhelical region. Although these data come from chimeric systems, they show direct potentiality for acquiring a membrane inserted state. Thus, the above fragments should be considered for the definition of plausible models of the active, membrane-bound species of Bcl-2 proteins - Curve-fitting overlapped bands: quantification and improvement of curve-fitting robustness in the presence of errors in the model and in the data, Lórenz-Fonfría VA & Padrós E.
The Analyst (2004)
129: 1243-1250.
Estimation of the band parameters of overlapped bands often relies on curve-fitting. It has been demonstrated that curve-fitting provides the maximum likelihood estimation of band parameters under a series of assumptions. One of these assumptions is that the curve-fitting model is correct and any error in the data is random. Under real conditions, we have to acknowledge the unavoidable presence of errors in the model and systematic errors in the data. Here, we derive an expression for the estimation of how these errors affect the quality of the parameters obtained from curve-fitting. In addition, we derive theoretical expressions to quantify the extent to which different methods can improve the curve-fitting robustness to these errors. The methods considered are: (i) deterministic and (ii) probabilistic constraints in the band parameters, (iii) curve-fitting band-narrowed data, and (iv) building a more accurate model. The theoretical expressions obtained are tested in the curve-fitting of a synthetic noisy spectrum with either baseline or band shape errors, and in the curve-fitting of the experimental infrared amide I band of the membrane protein bacteriorhodopsin - Curve-fitting of Fourier manipulated spectra comprising apodization, smoothing, derivation and deconvolution, Lórenz-Fonfría VA & Padrós E.
Spectrochim Acta A Mol Biomol Spectrosc (2004)
60: 2703-2710.
We present a general method for curve-fitting Fourier manipulated spectra, comprising apodized, smoothed, derivatised and deconvoluted spectra. The analytical expressions of Fourier manipulated bands in the spectral domain, needed for the curve-fitting, are usually very complex or do not even exist; hence an accurate curve-fit of Fourier manipulated spectra becomes unfeasible. Our strategy is to construct both the model and their derivatives in the Fourier domain, where they have simple and general expressions, and then Fourier transform them back to the spectral domain. The first benefit of this approach is the accurate curve-fitting of Fourier deconvoluted spectra, a main step in the secondary structure estimation of proteins by FTIR spectroscopy - Influence of proline residues in transmembrane helix packing, Orzáez M, Salgado J, Giménez-Giner A, Pérez-Payá E & Mingarro I.
J Mol Biol (2004)
335: 631-640.
Integral membrane proteins often contain proline residues in their alpha-helical transmembrane (TM) fragments, which may strongly influence their folding and association. Pro-scanning mutagenesis of the helical domain of glycophorin A (GpA) showed that replacement of the residues located at the center abrogates helix packing while substitution of the residues forming the ending helical turns allows dimer formation. Synthetic TM peptides revealed that a point mutation of one of the residues of the dimerization motif (L75P) located at the N-terminal helical turn of the GpA TM fragment, adopts a secondary structure and oligomeric state similar to the wild-type sequence in detergents. In addition, both glycosylation mapping in biological membranes and molecular dynamics showed that the presence of a proline residue at the lipid/water interface has as an effect the extension of the helical end. Thus, helix packing can be an important factor that determines appearance of proline in TM helices. Membrane proteins might accumulate proline residues at the two ends of their TM segments in order to modulate the exposition of key amino acid residues at the interface for molecular recognition events while allowing stable association and native folding - Structural and functional implications of the instability of the ADP/ATP transporter purified from mitochondria as revealed by FTIR spectroscopy, Lórenz-Fonfría VA, Villaverde J, Trézéguet V, Lauquin GJ-M, Brandolin G & Padrós E.
Biophys J (2003)
85: 255-266.
The ADP/ATP transporter shows a high instability when solubilized, making it difficult to obtain functional protein with sufficient purity for long-term spectroscopic studies. When solubilized in the detergent dodecyl maltoside the protein is in equilibrium between the so-called CATR and BA conformations and in a few hours it becomes nonfunctional, unable to bind either its inhibitors or its substrates. By Fourier transform infrared spectroscopy, we studied the structural changes involved in this denaturation process. To do so, the carboxyatractyloside-inhibited protein was used as a structural model for the protein in the CATR conformation and its spectrum was compared with that of the unliganded time-inactivated protein. From the difference spectra of the amide I, amide II, and amide A bands combined with dichroism spectra of the carboxyatractyloside-inhibited protein, we concluded that few structural differences exist between both states, affecting as few as 11 amino acids (3.5% of the protein); the structural changes consisted in the disappearance of large loop structure and the appearance of aggregated strands. We hypothesize that some mitochondrial loop (tentatively loop M1) shows a high tendency to aggregate, being responsible for the observed features. The functional consequences of this hypothesis are discussed
2001-02
- Study of amide-proton exchange of Escherichia coli melibiose permease by attenuated total reflection-Fourier transform infrared spectroscopy: evidence of structure modulation by substrate binding, Dave N, Lórenz-Fonfría VA, Villaverde J, Lemonnier R, Leblanc G & Padrós E.
J Biol Chem (2002)
277: 3380-3387.
The accessibility of Escherichia coli melibiose permease to aqueous solvent was studied following hydrogen-deuterium exchange kinetics monitored by attenuated total reflection-Fourier transform infrared spectroscopy under four distinct conditions where MelB forms different complexes with its substrates (H(+), Na(+), melibiose). Analysis of the amide II band upon (2)H(2)O exposure discloses a significant sugar protection of the protein against aqueous solvent, resulting in an 8% less exchange of the corresponding H(+)*melibiose*MelB complex compared with the protein in the absence of sugar. Investigation of the amide I exchange reveals clear substrate effects on beta-sheet accessibility, with the complex H(+)*melibiose*MelB being the most protected state against exchange, followed by Na(+)*melibiose*MelB. Although of smaller magnitude, similar changes in alpha-helices plus non-ordered structures are detected. Finally, no differences are observed when analyzing reverse turn structures. The results suggest that sugar binding induces a remarkable compactness of the carrier's structure, affecting mainly beta-sheet domains of the transporter, which, according to secondary structure predictions, may include cytoplasmic loops 4-5 and 10-11. A possible catalytic role of these two loops in the functioning of MelB is hypothesized - Fourier Deconvolution in Non-Self-Deconvolving Conditions. Effective Narrowing, Signal-to-Noise Degradation, and Curve Fitting, Lórenz-Fonfría VA, Villaverde J & Padrós E.
Appl Spectrosc (2002)
56: 232-242.
The effect of Fourier deconvolution on band narrowing and on the decrease of signal-to-noise ratio has been studied for a generalized case in which the width used in the deconvolution does not match the actual bandwidth. For the identification of underlying component bands, our results show that application of infra-deconvolution (i.e., the bandwidth used for deconvolution is lower than the actual bandwidth) produces a high degradation of the signal-to-noise ratio and poor band narrowing. On the contrary, self- or over-deconvolution, with lower signal-to-noise degradation and higher band narrowing, seem more suitable for this purpose. Relative to quantitative analysis, we rely on both theoretical and practical aspects to propose the generalized use of Voigt band shapes as a fairly correct general model to be used in the curve fitting of deconvoluted bands. With its use, the curve fitting of noise-free deconvoluted bands retrieved the original band parameters with high accuracy. The noise effect on the parameter precision obtained by curve fitting a deconvoluted noisy Lorentzian band was also studied. Finally, the existence of optimum deconvolution parameters for curve fitting complex spectra is considered, and a general recommendation for approaching this optimum is given. - Peptides in apoptosis research, Salgado J, García-Sáez AJ, Malet G, Mingarro I & Pérez-Payá E.
J Pept Sci (2002)
8: 543-560.
Apoptosis is a complex process that plays a central role in physiological and pathological cell death. This fast evolving research area has experienced incredible development in the past few years. Progress in the knowledge of the structure of many of the main molecular actors of the apoptotic signal transduction pathways has driven the design of synthetic peptides that in some cases can function as simplified versions of their parent proteins. These molecules are contributing to a better understanding of the activity and regulation of apoptotic proteins and also are setting the basis for the discovery of effective drugs to combat important diseases related to apoptosis. Most applications of peptides in apoptosis research are so far related to caspases, caspase regulatory proteins, such as LAPs and Smac, and proteins of the Bcl-2 family. Additionally, important perspectives are open to other systems, such as the macromolecular assemblies that are responsible for the activation of initiator caspases - Solid state 19F-NMR of biomembranes, Grage SL, Salgado J, Dürr U, Afonin S, Glaser RW & Ulrich AS.
In: Perspectives on Solid State NMR in Biology. Focus on Structural Biology, Vol. 1, Kiihne, S.R. and de Groot, H.J.M.
(Ed.). Springer, Dordrecht, 2001,
pp. 83-91.
Structure determination of membrane-associated polypeptides presents one of the major challenges to solid state NMR spectroscopy. Many studies have been carried out so far using selective isotope labels, such as 2H, 13C, or 15N. These NMR-reporters can be incorporated into the protein backbone or side chains, to reveal local structural parameters and to describe the dynamic properties of the membrane-embedded molecule. For example, an distance r can be measured between a pair of labels by means of dipolar recoupling MAS techniques such as rotational resonance or REDOR. Alternatively, uniaxially oriented samples are used to determine the angle A of a labelled molecular segment with respect to the membrane normal N. The latter approach relies on the orientation-dependent resonance frequency, which carries information about the anisotropie chemical shift tensor (13C, 15N), the dipolar coupling (1H-15N), or the quadrupolar interaction (2H) - The Secondary Structure of the Inhibited Mitochondrial ADP/ATP Transporter from Yeast Analyzed by FTIR Spectroscopytextdagger, Lórenz VA, Villaverde J, Trézéguet V, Lauquin GJ-M, Brandolin G & Padrós E.
Biochemistry (2001)
40: 8821-8833.
Fourier transform infrared spectroscopy has been applied to the study of the carboxyatractyloside-inhibited mitochondrial ADP/ATP transporter from the yeast Saccharomyces cerevisiae, either solubilized in dodecyl maltoside or reconstituted in phosphatidylcholine liposomes. Its secondary structure has been estimated by means of Fourier self-deconvolution followed by curve fit. A Voigt function was used to fit the components of the deconvoluted spectrum, aiming to account for any distortions introduced by deconvolution. For any of the states analyzed, reconstituted or solubilized, in solution or in dry films, 60-70% of the amino acids are found to adopt alpha-helix plus unordered structures, coherent with the six transmembrane spanning helix model. Moreover, the problem of structure preservation on drying was addressed, and several observations pointed to a maintenance of the protein structure in dry films. Comparison of reconstituted and solubilized samples indicated the presence of both lipid-induced changes in the protein (decrease of the beta-sheets and increase of unordered structures) and protein-induced changes in the lipids (strong hydrogen bonding of lipid C=O groups). To obtain a better discrimination of alpha-helix and unordered structure contributions for the reconstituted form, H/D exchange experiments were performed. Between 35% and 45% of the amino acids were finally assigned to alpha-helix structures, compatible with the existence of five or six transmembrane spanning helices in the transporter. The level of H/D exchange was determined after 15 h of exposure to D(2)O vapor to be 85%, reflecting a high accessibility of the amide hydrogens even for the carboxyatractyloside-inhibited state. - Membrane-bound structure and alignment of the antimicrobial beta-sheet peptide gramicidin S derived from angular and distance constraints by solid state 19F-NMR, Salgado J, Grage SL, Kondejewski LH, Hodges RS, McElhaney RN & Ulrich AS.
J Biomol NMR (2001)
21: 191-208.
The antimicrobial properties of the cyclic beta-sheet peptide gramicidin S are attributed to its destabilizing effect on lipid membranes. Here we present the membrane-bound structure and alignment of a derivative of this peptide, based on angular and distance constraints. Solid-state 19F-NMR was used to study a 19F-labelled gramicidin S analogue in dimyristoylphosphatidylcholine bilayers at a lipid:peptide ratio of 80:1 and above. Two equivalent leucine side chains were replaced by the non-natural amino acid 4F-phenylglycine, which serves as a highly sensitive reporter on the structure and dynamics of the peptide backbone. Using a modified CPMG multipulse sequence, the distance between the two 19F-labels was measured from their homonuclear dipolar coupling as 6 A. in good agreement with the known backbone structure of natural gramicidin S in solution. By analyzing the anisotropic chemical shift of the 19F-labels in macroscopically oriented membrane samples, we determined the alignment of the peptide in the bilayer and described its temperature-dependent mobility. In the gel phase, the 19F-labelled gramicidin S is aligned symmetrically with respect to the membrane normal, i.e., with its cyclic beta-sheet backbone lying flat in the plane of the bilayer, which is fully consistent with its amphiphilic character. Upon raising the temperature to the liquid crystalline state, a considerable narrowing of the 19F-NMR chemical shift dispersion is observed, which is attributed the onset of global rotation of the peptide and further wobbling motions. This study demonstrates the potential of the 19F nucleus to describe suitably labelled polypeptides in membranes, requiring only little material and short NMR acquisition times

