
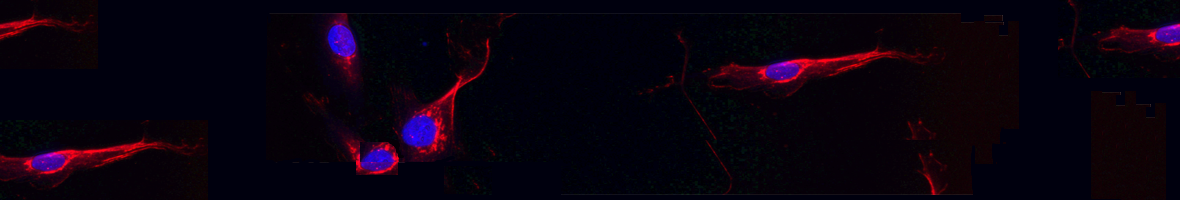
-
Proof of concept of peptide-linked blockmiR-induced MBNL functional rescue in myotonic dystrophy type 1 mouse model
Overby SJ, Cerro-Herreros E, González-Martínez I, Varela MA, Seoane-Miraz D, Jad Y, Raz R, Møller T, Pérez-Alonso M, Wood MJ, Llamusí B, Artero R - 2022 - Mol Ther Nucleic Acids
(2022).ArticleMyotonic dystrophy type 1 is a debilitating neuromuscular disease causing muscle weakness, myotonia, and cardiac dysfunction. The phenotypes are caused by muscleblind-like (MBNL) protein sequestration by toxic RNA in the DM1 protein kinase (DMPK) gene. DM1 patients exhibit a pathogenic number of repetitions in DMPK, which leads to downstream symptoms. Another disease characteristic is altered microRNA (miRNA) expression. It was previously shown that miR-23b regulates the translation of MBNL1 into protein. Antisense oligonucleotide (AON) treatment targeting this miRNA can improve disease symptoms. Here, we present a refinement of this strategy targeting a miR-23b binding site on the MBNL1 3'...
Myotonic dystrophy type 1 is a debilitating neuromuscular disease causing muscle weakness, myotonia, and cardiac dysfunction. The phenotypes are caused by muscleblind-like (MBNL) protein sequestration by toxic RNA in the DM1 protein kinase (DMPK) gene. DM1 patients exhibit a pathogenic number of repetitions in DMPK, which leads to downstream symptoms. Another disease characteristic is altered microRNA (miRNA) expression. It was previously shown that miR-23b regulates the translation of MBNL1 into protein. Antisense oligonucleotide (AON) treatment targeting this miRNA can improve disease symptoms. Here, we present a refinement of this strategy targeting a miR-23b binding site on the MBNL1 3' UTR in DM1 model cells and mice by using AONs called blockmiRs. BlockmiRs linked to novel cell-penetrating peptide chemistry showed an increase in MBNL1 protein in DM1 model cells and HSALR mice. They also showed an increase in muscle strength and significant rescue of downstream splicing and histological phenotypes in mice without disturbing the endogenous levels of other miR-23b target transcripts.
Llegir mésOcultar DOI: 10.1016/j.omtn.2022.02.003 -
Deciphering the Complex Molecular Pathogenesis of Myotonic Dystrophy Type 1 through Omics Studies
Espinosa-Espinosa J, González-Barriga A, López-Castel A, Artero R - 2022 - Int J Mol Sci
(2022).ArticleOmics studies are crucial to improve our understanding of myotonic dystrophy type 1 (DM1), the most common muscular dystrophy in adults. Employing tissue samples and cell lines derived from patients and animal models, omics approaches have revealed the myriad alterations in gene and microRNA expression, alternative splicing, 3' polyadenylation, CpG methylation, and proteins levels, among others, that contribute to this complex multisystem disease. In addition, omics characterization of drug candidate treatment experiments provides crucial insight into the degree of therapeutic rescue and off-target effects that can be achieved. Finally, several innovative technologies such as single-cell...
Omics studies are crucial to improve our understanding of myotonic dystrophy type 1 (DM1), the most common muscular dystrophy in adults. Employing tissue samples and cell lines derived from patients and animal models, omics approaches have revealed the myriad alterations in gene and microRNA expression, alternative splicing, 3' polyadenylation, CpG methylation, and proteins levels, among others, that contribute to this complex multisystem disease. In addition, omics characterization of drug candidate treatment experiments provides crucial insight into the degree of therapeutic rescue and off-target effects that can be achieved. Finally, several innovative technologies such as single-cell sequencing and artificial intelligence will have a significant impact on future DM1 research.
Llegir mésOcultar DOI: 10.3390/ijms23031441 -
Inhibition of autophagy rescues muscle atrophy in a LGMDD2 Drosophila model
Blázquez-Bernal Á, Fernandez-Costa JM, Bargiela A, Artero R - 2021 - FASEB J
(2021).ArticleLimb-girdle muscular dystrophy D2 (LGMDD2) is an ultrarare autosomal dominant myopathy caused by mutation of the normal stop codon of the TNPO3 nuclear importin. The mutant protein carries a 15 amino acid C-terminal extension associated with pathogenicity. Here we report the first animal model of the disease by expressing the human mutant TNPO3 gene in Drosophila musculature or motor neurons and concomitantly silencing the endogenous expression of the fly protein ortholog. A similar genotype expressing wildtype TNPO3 served as a control. Phenotypes characterization revealed that mutant TNPO3 expression targeted at muscles or motor neurons caused LGMDD2-like phenotypes such as muscle...
Limb-girdle muscular dystrophy D2 (LGMDD2) is an ultrarare autosomal dominant myopathy caused by mutation of the normal stop codon of the TNPO3 nuclear importin. The mutant protein carries a 15 amino acid C-terminal extension associated with pathogenicity. Here we report the first animal model of the disease by expressing the human mutant TNPO3 gene in Drosophila musculature or motor neurons and concomitantly silencing the endogenous expression of the fly protein ortholog. A similar genotype expressing wildtype TNPO3 served as a control. Phenotypes characterization revealed that mutant TNPO3 expression targeted at muscles or motor neurons caused LGMDD2-like phenotypes such as muscle degeneration and atrophy, and reduced locomotor ability. Notably, LGMDD2 mutation increase TNPO3 at the transcript and protein level in the Drosophila model Upregulated muscle autophagy observed in LGMDD2 patients was also confirmed in the fly model, in which the anti-autophagic drug chloroquine was able to rescue histologic and functional phenotypes. Overall, we provide a proof of concept of autophagy as a target to treat disease phenotypes and propose a neurogenic component to explain mutant TNPO3 pathogenicity in diseased muscles.
Llegir mésOcultar DOI: 10.1096/fj.202100539RR -
Defined D-hexapeptides bind CUG repeats and rescue phenotypes of myotonic dystrophy myotubes in a Drosophila model of the disease
Rapisarda A, Bargiela A, Llamusi B, Pont I, Estrada-Tejedor R, Garcia-España E, Artero R, Perez-Alonso M - 2021 - Sci Rep
(2021).ArticleIn Myotonic Dystrophy type 1 (DM1), a non-coding CTG repeats rare expansion disease; toxic double-stranded RNA hairpins sequester the RNA-binding proteins Muscleblind-like 1 and 2 (MBNL1 and 2) and trigger other DM1-related pathogenesis pathway defects. In this paper, we characterize four D-amino acid hexapeptides identified together with abp1, a peptide previously shown to stabilize CUG RNA in its single-stranded conformation. With the generalized sequence cpy(a/t)(q/w)e, these related peptides improved three MBNL-regulated exon inclusions in DM1-derived cells. Subsequent experiments showed that these compounds generally increased the relative expression of MBNL1 and its...
In Myotonic Dystrophy type 1 (DM1), a non-coding CTG repeats rare expansion disease; toxic double-stranded RNA hairpins sequester the RNA-binding proteins Muscleblind-like 1 and 2 (MBNL1 and 2) and trigger other DM1-related pathogenesis pathway defects. In this paper, we characterize four D-amino acid hexapeptides identified together with abp1, a peptide previously shown to stabilize CUG RNA in its single-stranded conformation. With the generalized sequence cpy(a/t)(q/w)e, these related peptides improved three MBNL-regulated exon inclusions in DM1-derived cells. Subsequent experiments showed that these compounds generally increased the relative expression of MBNL1 and its nuclear-cytoplasmic distribution, reduced hyperactivated autophagy, and increased the percentage of differentiated (Desmin-positive) cells in vitro. All peptides rescued atrophy of indirect flight muscles in a Drosophila model of the disease, and partially rescued muscle function according to climbing and flight tests. Investigation of their mechanism of action supports that all four compounds can bind to CUG repeats with slightly different association constant, but binding did not strongly influence the secondary structure of the toxic RNA in contrast to abp1. Finally, molecular modeling suggests a detailed view of the interactions of peptide-CUG RNA complexes useful in the chemical optimization of compounds.
Llegir mésOcultar DOI: 10.1038/s41598-021-98866-0 -
Practicing logical reasoning through Drosophila segmentation gene mutants
Bargiela A, Artero R - 2021 - Biochem Mol Biol Educ
(2021).ArticleLaboratory practical sessions are critical to scientific training in biology but usually fail to promote logical and hypothesis-driven reasoning and rely heavily on the teacher's instructions. This paper describes a 2-day laboratory practicum in which students prepare and analyze larval cuticle preparations of Drosophila segmentation gene mutant strains. Embryonic segmentation involves three major classes of genes according to their loss-of-function phenotypes: the establishment of broad regions by gap genes, the specification of the segments by the pair-rule genes, and the compartments within segments by the segment polarity genes. Students are asked to sort undefined segmentation mutants...
Laboratory practical sessions are critical to scientific training in biology but usually fail to promote logical and hypothesis-driven reasoning and rely heavily on the teacher's instructions. This paper describes a 2-day laboratory practicum in which students prepare and analyze larval cuticle preparations of Drosophila segmentation gene mutant strains. Embryonic segmentation involves three major classes of genes according to their loss-of-function phenotypes: the establishment of broad regions by gap genes, the specification of the segments by the pair-rule genes, and the compartments within segments by the segment polarity genes. Students are asked to sort undefined segmentation mutants into gap, pair-rule, or segment polarity categories based on their knowledge of the Drosophila segmentation process and the microscopic anatomical traits they are capable of finding in the sample preparations. This technically simple practicum prompts students to pay attention to detailed observation to detect anatomic markers of intrasegmental compartments and thorax versus abdomen cuticle, and promote their logical reasoning in hypothesizing to which segmentation type a given mutant fits best.
Llegir mésOcultar DOI: 10.1002/bmb.21554 -
Musashi-2 contributes to myotonic dystrophy muscle dysfunction by promoting excessive autophagy through miR-7 biogenesis repression
Sabater-Arcis M, Bargiela A, Moreno N, Poyatos-Garcia J, Vilchez JJ, Artero R - 2021 - Mol Ther Nucleic Acids
(2021).ArticleSkeletal muscle symptoms strongly contribute to mortality of myotonic dystrophy type 1 (DM1) patients. DM1 is a neuromuscular genetic disease caused by CTG repeat expansions that, upon transcription, sequester the Muscleblind-like family of proteins and dysregulate alternative splicing of hundreds of genes. However, mis-splicing does not satisfactorily explain muscle atrophy and wasting, and several other contributing factors have been suggested, including hyperactivated autophagy leading to excessive catabolism. MicroRNA (miR)-7 has been demonstrated to be necessary and sufficient to repress the autophagy pathway in cell models of the disease, but the origin of its low levels in DM1 was...
Skeletal muscle symptoms strongly contribute to mortality of myotonic dystrophy type 1 (DM1) patients. DM1 is a neuromuscular genetic disease caused by CTG repeat expansions that, upon transcription, sequester the Muscleblind-like family of proteins and dysregulate alternative splicing of hundreds of genes. However, mis-splicing does not satisfactorily explain muscle atrophy and wasting, and several other contributing factors have been suggested, including hyperactivated autophagy leading to excessive catabolism. MicroRNA (miR)-7 has been demonstrated to be necessary and sufficient to repress the autophagy pathway in cell models of the disease, but the origin of its low levels in DM1 was unknown. We have found that the RNA-binding protein Musashi-2 (MSI2) is upregulated in patient-derived myoblasts and biopsy samples. Because it has been previously reported that MSI2 controls miR-7 biogenesis, we tested the hypothesis that excessive MSI2 was repressing miR-7 maturation. Using gene-silencing strategies (small interfering RNAs [siRNAs] and gapmers) and the small molecule MSI2-inhibitor Ro 08-2750, we demonstrate that reducing MSI2 levels or activity boosts miR-7 expression, represses excessive autophagy, and downregulates atrophy-related genes of the UPS system. We also detect a significant upregulation of MBNL1 upon MSI2 silencing. Taken together, we propose MSI2 as a new therapeutic target to treat muscle dysfunction in DM1.
Llegir mésOcultar DOI: 10.1016/j.omtn.2021.08.010 -
Preclinical characterization of antagomiR-218 as a potential treatment for myotonic dystrophy
Cerro-Herreros E, González-Martínez I, Moreno N, Espinosa-Espinosa J, Fernández-Costa JM, Colom-Rodrigo A, Overby SJ, Seoane-Miraz D, Poyatos-García J, Vilchez JJ, López de Munain A, Varela MA, Wood MJ, Pérez-Alonso M, Llamusí B, Artero R - 2021 - Mol Ther Nucleic Acids
(2021).ArticleMyotonic dystrophy type 1 (DM1) is a rare neuromuscular disease caused by expansion of unstable CTG repeats in a non-coding region of the DMPK gene. CUG expansions in mutant DMPK transcripts sequester MBNL1 proteins in ribonuclear foci. Depletion of this protein is a primary contributor to disease symptoms such as muscle weakness and atrophy and myotonia, yet upregulation of endogenous MBNL1 levels may compensate for this sequestration. Having previously demonstrated that antisense oligonucleotides against miR-218 boost MBNL1 expression and rescue phenotypes in disease models, here we provide preclinical characterization of an antagomiR-218 molecule using the HSALR mouse model and...
Myotonic dystrophy type 1 (DM1) is a rare neuromuscular disease caused by expansion of unstable CTG repeats in a non-coding region of the DMPK gene. CUG expansions in mutant DMPK transcripts sequester MBNL1 proteins in ribonuclear foci. Depletion of this protein is a primary contributor to disease symptoms such as muscle weakness and atrophy and myotonia, yet upregulation of endogenous MBNL1 levels may compensate for this sequestration. Having previously demonstrated that antisense oligonucleotides against miR-218 boost MBNL1 expression and rescue phenotypes in disease models, here we provide preclinical characterization of an antagomiR-218 molecule using the HSALR mouse model and patient-derived myotubes. In HSALR, antagomiR-218 reached 40-60 pM 2 weeks after injection, rescued molecular and functional phenotypes in a dose- and time-dependent manner, and showed a good toxicity profile after a single subcutaneous administration. In muscle tissue, antagomiR rescued the normal subcellular distribution of Mbnl1 and did not alter the proportion of myonuclei containing CUG foci. In patient-derived cells, antagomiR-218 improved defective fusion and differentiation and rescued up to 34% of the gene expression alterations found in the transcriptome of patient cells. Importantly, miR-218 was found to be upregulated in DM1 muscle biopsies, pinpointing this microRNA (miRNA) as a relevant therapeutic target.
Llegir mésOcultar DOI: 10.1016/j.omtn.2021.07.017 -
Rabphilin silencing causes dilated cardiomyopathy in a Drosophila model of nephrocyte damage
Selma-Soriano E, Casillas-Serra C, Artero R, Llamusi B, Navarro JA, Redón J - 2021 - Sci Rep
(2021).ArticleHeart failure (HF) and the development of chronic kidney disease (CKD) have a direct association. Both can be cause and consequence of the other. Many factors are known, such as diabetes or hypertension, which can lead to the appearance and/or development of these two conditions. However, it is suspected that other factors, namely genetic ones, may explain the differences in the manifestation and progression of HF and CKD among patients. One candidate factor is Rph, a gene expressed in the nervous and excretory system in mammals and Drosophila, encoding a Rab small GTPase family effector protein implicated in vesicular trafficking. We found that Rph is expressed in the Drosophila heart, and...
Heart failure (HF) and the development of chronic kidney disease (CKD) have a direct association. Both can be cause and consequence of the other. Many factors are known, such as diabetes or hypertension, which can lead to the appearance and/or development of these two conditions. However, it is suspected that other factors, namely genetic ones, may explain the differences in the manifestation and progression of HF and CKD among patients. One candidate factor is Rph, a gene expressed in the nervous and excretory system in mammals and Drosophila, encoding a Rab small GTPase family effector protein implicated in vesicular trafficking. We found that Rph is expressed in the Drosophila heart, and the silencing of Rph gene expression in this organ had a strong impact in the organization of fibers and functional cardiac parameters. Specifically, we observed a significant increase in diastolic and systolic diameters of the heart tube, which is a phenotype that resembles dilated cardiomyopathy in humans. Importantly, we also show that silencing of Rabphilin (Rph) expression exclusively in the pericardial nephrocytes, which are part of the flies' excretory system, brings about a non-cell-autonomous effect on the Drosophila cardiac system. In summary, in this work, we demonstrate the importance of Rph in the fly cardiac system and how silencing Rph expression in nephrocytes affects the Drosophila cardiac system.
Llegir mésOcultar DOI: 10.1038/s41598-021-94710-7 -
Myotonic dystrophy type 1 drug development: A pipeline toward the market
Pascual-Gilabert M, López-Castel A, Artero R - 2021 - Drug Discov Today
(2021).ArticleMyotonic dystrophy type 1 (DM1) is a multisystemic neuromuscular genetic disease with an estimated prevalence of approximately at least half a million individuals based on its vast ethnic variation. Building upon a well-known physiopathology and several proof-of-concept therapeutic approaches, herein we compile a comprehensive overview of the most recent drug development programs under preclinical and clinical evaluation. Specifically, close to two dozen drug developments, eight of which are already in clinical trials, explore a diversity of new chemical entities, drug repurposing, oligonucleotide, and gene therapy-based approaches. Of these, repurposing of tideglusib, mexiletine, or...
Myotonic dystrophy type 1 (DM1) is a multisystemic neuromuscular genetic disease with an estimated prevalence of approximately at least half a million individuals based on its vast ethnic variation. Building upon a well-known physiopathology and several proof-of-concept therapeutic approaches, herein we compile a comprehensive overview of the most recent drug development programs under preclinical and clinical evaluation. Specifically, close to two dozen drug developments, eight of which are already in clinical trials, explore a diversity of new chemical entities, drug repurposing, oligonucleotide, and gene therapy-based approaches. Of these, repurposing of tideglusib, mexiletine, or metformin appear to be therapies with the most potential to receive marketing authorization for DM1.
Llegir mésOcultar DOI: 10.1016/j.drudis.2021.03.024 -
The hallmarks of myotonic dystrophy type 1 muscle dysfunction
Ozimski LL, Sabater-Arcis M, Bargiela A, Artero R - 2021 - Biol Rev Camb Philos Soc
(2021).ArticleMyotonic dystrophy type 1 (DM1) is the most prevalent form of muscular dystrophy in adults and yet there are currently no treatment options. Although this disease causes multisystemic symptoms, it is mainly characterised by myopathy or diseased muscles, which includes muscle weakness, atrophy, and myotonia, severely affecting the lives of patients worldwide. On a molecular level, DM1 is caused by an expansion of CTG repeats in the 3' untranslated region (3'UTR) of the DM1 Protein Kinase (DMPK) gene which become pathogenic when transcribed into RNA forming ribonuclear foci comprised of auto complementary CUG hairpin structures that can bind proteins. This leads to the sequestration of the...
Myotonic dystrophy type 1 (DM1) is the most prevalent form of muscular dystrophy in adults and yet there are currently no treatment options. Although this disease causes multisystemic symptoms, it is mainly characterised by myopathy or diseased muscles, which includes muscle weakness, atrophy, and myotonia, severely affecting the lives of patients worldwide. On a molecular level, DM1 is caused by an expansion of CTG repeats in the 3' untranslated region (3'UTR) of the DM1 Protein Kinase (DMPK) gene which become pathogenic when transcribed into RNA forming ribonuclear foci comprised of auto complementary CUG hairpin structures that can bind proteins. This leads to the sequestration of the muscleblind-like (MBNL) family of proteins, depleting them, and the abnormal stabilisation of CUGBP Elav-like family member 1 (CELF1), enhancing it. Traditionally, DM1 research has focused on this RNA toxicity and how it alters MBNL and CELF1 functions as key splicing regulators. However, other proteins are affected by the toxic DMPK RNA and there is strong evidence that supports various signalling cascades playing an important role in DM1 pathogenesis. Specifically, the impairment of protein kinase B (AKT) signalling in DM1 increases autophagy, apoptosis, and ubiquitin-proteasome activity, which may also be affected in DM1 by AMP-activated protein kinase (AMPK) downregulation. AKT also regulates CELF1 directly, by affecting its subcellular localisation, and indirectly as it inhibits glycogen synthase kinase 3 beta (GSK3β), which stabilises the repressive form of CELF1 in DM1. Another kinase that contributes to CELF1 mis-regulation, in this case by hyperphosphorylation, is protein kinase C (PKC). Additionally, it has been demonstrated that fibroblast growth factor-inducible 14 (Fn14) is induced in DM1 and is associated with downstream signalling through the nuclear factor κB (NFκB) pathways, associating inflammation with this disease. Furthermore, MBNL1 and CELF1 play a role in cytoplasmic processes involved in DM1 myopathy, altering proteostasis and sarcomere structure. Finally, there are many other elements that could contribute to the muscular phenotype in DM1 such as alterations to satellite cells, non-coding RNA metabolism, calcium dysregulation, and repeat-associated non-ATG (RAN) translation. This review aims to organise the currently dispersed knowledge on the different pathways affected in DM1 and discusses the unexplored connections that could potentially help in providing new therapeutic targets in DM1 research.
Llegir mésOcultar DOI: 10.1111/brv.12674
 Institut Universitari de Biotecnologia i Biomedicina (BIOTECMED)
Institut Universitari de Biotecnologia i Biomedicina (BIOTECMED)


© 2026 UV. - Av. Vicent Andrés Estellés, 19. 46100. Burjassot. València. Espanya. Tel. +34 963 543 460