ENFERMEDADES AMPOLLOSAS AUTOINMUNES
Semiológicamente definimos
una ampolla como una lesión primaria llena de liquido de más
de 1 cm. y una vesícula cuando es de menor tamaño, a pesar
de esta definición de ampollas el espectro de enfermedades ampollosas
es más amplio, clasificándose dentro de las enfermedades
ampollosas aquellas entidades que se caracterizan por la alteración
en la cohesión entre las estructuras cutáneas independientemente
de que se expresen clínicamente como ampollas, vesículas
u otro tipo de lesiones clínicas.
Las ampollas pueden desarrollarse
en piel y mucosas, pueden ser localizadas o generalizadas y localizarse
en cualquier nivel cutáneo (epidérmico, unión dermo-epidérmica,
dérmica). Las ampollas pueden tener diferentes causas (fig 1), pueden
ser congénitas (por alteración congénita en las estructuras
de cohesión) o adquiridas. Las adquiridas pueden tener a su vez
diferentes orígenes: físicas, infecciosas, isquémicas,
metabólicas, autoinmunes, etc.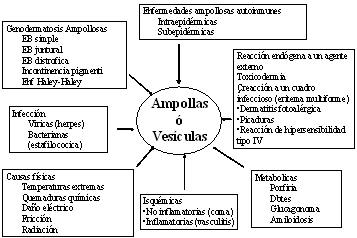 El
objetivo de este tema va a ser estudiar las enfermedades ampollosas autoinmunes
considerándose como tales a aquellas enfermedades caracterizadas por la
presencia de autoanticuerpos patogénicos dirigidos contra antígenos
funcionales de la unión intercelular o de la unión del epitelio a la dermis. El
diagnóstico de estas enfermedades ampollosas autoinmunes requiere el
conocimiento de las manifestaciones clínicas, de los cambios
histopatológicos y también requiere de la demostración de los anticuerpos
bien en los tejidos (inmunofluorescencia directa) o circulantes
(inmunofluorescencia indirecta y técnicas de ELISA y Western-blot). En este
tema estudiaremos los siguientes apartados
El
objetivo de este tema va a ser estudiar las enfermedades ampollosas autoinmunes
considerándose como tales a aquellas enfermedades caracterizadas por la
presencia de autoanticuerpos patogénicos dirigidos contra antígenos
funcionales de la unión intercelular o de la unión del epitelio a la dermis. El
diagnóstico de estas enfermedades ampollosas autoinmunes requiere el
conocimiento de las manifestaciones clínicas, de los cambios
histopatológicos y también requiere de la demostración de los anticuerpos
bien en los tejidos (inmunofluorescencia directa) o circulantes
(inmunofluorescencia indirecta y técnicas de ELISA y Western-blot). En este
tema estudiaremos los siguientes apartados
-
Conocer las estructuras de adhesión cutánea
-
Conocer los métodos de estudio utilizados en las enfermedades ampollosas
autoinmunes
-
Conocer las características de las entidades
-
Conocer los tratamientos utilizados.
Estructuras de adhesión
En la actualidad se conoce con bastante detalle las estructuras y proteínas
involucradas en mantener intacta la cohesión cutánea, cuando
estas estructuras se ven alteradas es cuando se producen las ampollas.
Estas estructuras están constituidas principalmente por los desmosomas
-que mantienen la unión entre los queratinocitos- y los hemidesmosomas
-que mantienen la unión entre los queratinocitos basales y la membrana
basal.
Los desmosomas son las estructuras que mantienen la unión intercelular,
consisten en 2 placas citoplásmicas, simétricas, de 2 células
adyacentes y de un núcleo central (desmoglea) que tiene un grosor
de unos 30 nm y que se sitúa entre las 2 placas desmosomales. Las
placas desmosomales se localizan en la porción interna de la membrana
citoplásmica, es una región electrodensa de 14-20 nm de grosor,
en la que se insertan los filamentos intermedios de queratina del citoplasma. Dado que la queratina
no atraviesa la membrana celular, para mantener la unión intercelular
existen unas proteínas que cumplen esa función. Así
las queratinas se unen a unas proteínas de la placa desmosomal (plaquinas)
que a su vez se unen a unas proteínas de membrana -cadherinas (desmogleina
1,2 y 3; desmocolina 1,2 y 3; cadherina). Las cadherinas de una célula
se unen al complejo cadherina-plaquina-queratina de la célula contigua.
La integridad de los desmosomas determina la adhesión epidérmica,
su alteración en genodermatosis o en enfermedades ampollosas autoinmunes
da lugar al desarrollo de enfermedades ampollosas.
La
unión dermo-epidermica
es también de vital importancia para mantener la cohesión
de la epidermis a la dermis. Esta unión dermo-epidermica está
constituida por varias estructuras que incluyen los hemidesmosomas,
los filamentos de anclaje, la lámina densa y las fibras de anclaje.
Los hemidesmosomas son estructuras localizadas en la porción basal
de la célula basal y actúan como puente entre el citoesqueleto
y la membrana basal, realizándose esta función por medio
de los filamentos de anclaje que son estructuras filamentosas que atraviesan
la lámina lúcida para insertarse en la lámina densa,
uniendo los hemidesmosomas a la membrana basal. Las fibras de anclaje se
originan en la lámina densa, introduciéndose en la dermis
papilar donde bien pueden insertarse en las placas de anclaje o retroceder
y volver a insertarse en la lámina densa. Estas estructuras están
constituidas por diversas proteínas que pueden comportarse como
antígenos en enfermedades ampollosas autoinmunes, así, a
nivel de los hemidesmosomas se encuentra la proteína BP230 (proteína
citoplásmica o antígeno del penfigoide ampolloso tipo I),
a nivel de la lámina lúcida se encuentra la proteína
BP180 (proteína de transmembrana o antígeno del penfigoide
ampolloso tipo II) y la laminina 5. La lámina densa está
constituida por colágeno tipo IV y las fibras de anclaje están
constituidas por colágeno tipo VII.
Métodos de estudio de las enfermedades ampollosas autoinmunes:
Para el diagnóstico
y estudio de las enfermedades ampollosas autoinmunes es necesario utilizar
un conjunto de métodos que incluyen datos clínicos, histológicos,
inmunológicos y los obtenidos en microscopía electrónica,
inmunoblot e inmunoprecipitación. La utilización sistemática
de los diferentes métodos diagnósticos permite conocer y
diferenciar las diferentes enfermedades ampollosas.
1.Clínica:
Diversos hallazgos clínicos
van a ser importantes en el estudio de las enfermedades ampollosas autoinmunes
incluyendo la edad, la asociación a enfermedades sistémicas,
los síntomas y la morfología de las lesiones.
Prácticamente todas las
enfermedades ampollosas autoinmunes se han descrito en cualquier edad,
sin embargo existe una mayor frecuencia de afectación en cada grupo
de edad, así la enfermedad ampollosas IgA lineal afecta predominantemente
a la infancia, la dermatitis herpetiforme y el penfigoide gestationis afecta
a adultos jóvenes, el pénfigo vulgar en la edad adulta y
el penfigoide tiene su máxima incidencia alrededor de los 80 años.
Morfológicamente vamos
a poder diferenciar entre las enfermedades ampollosas intraepidérmicas
y subepidérmicas. Las intraepidérmicas se caracterizan por
ser ampollas flácidas, de corta duración, observándose
frecuentemente erosiones y costras. Es característico de las enfermedades
ampollosas intraepidérmicas presentar un signo de Nikolsky
positivo. Las enfermedades ampollosas subepidérmicas se caracterizan
por desarrollar ampollas tensas, de contenido seroso, que suelen durar
más de 24 horas y que nosotros podremos objetivar con facilidad,
siendo en estos casos el signo de Nikolsky negativo. La afectación
de mucosas es un hallazgo importante siendo llamativo en el pénfigo
vulgar y en el penfigoide de mucosas y ausentes en en pénfigo foliáceo.
La distribución de las lesiones también es característica
de cada enfermedad, así el pénfigo vulgar suele iniciarse
por la afectación oral, las lesiones de la dermatitis herpetiforme
tienen una distribución simétrica afectando a caras extensoras
de extremidades y el pénfigiode ampolloso suele afectar a
pliegues. Los síntomas como el prurito si bien son frecuentes en
todas las enfermedades ampollosas son especialmente llamativas en la dermatitis
herpetiforme.
Algunas enfermedades ampollosas
tienen unas asociaciones claras como la dermatitis herpetiforme con la
enfermedad celíaca, el penfigoide gestationes con el embarazo o
período postparto, la epidermolisis ampollosa adquirida con enfermedad
inflamatoria intestinal y el pénfigo paraneoplásico
con neoplasias de origen hematológico.
3.Histología:
La realización de biopsias
cutáneas es imprescindible para el estudio de las enfermedades ampollosas.
Las biopsias deben realizarse en ampollas de reciente aparición,
de menos de 24-48 horas. Las biopsias deben incluir los 2 márgenes
laterales de la ampolla así como piel sana perilesional. Histológicamente
vamos a poder observar la localización de la ampolla, si se produce
en la epidermis (intraepidérmica) o por debajo de la epidermis (subepidérmica).
En las ampollas intraepidérmicas podremos observar la existencia
o ausencia de acantolisis, balonización o espongiosis así
como la existencia de infiltrado y características del infiltrado
inflamatorio que nos ayudaran a clasificar la enfermedad ampollosa. En
las ampollas subepidérmicas la observación del tipo de infiltrado
inflamatorio y su disposición nos serán útiles en
el mismo sentido.
4.Inmunofluorescencia:
Para establecer el diagnóstico
diferencial de las enfermedades ampollosas, se realizan 3 técnicas
de inmunofluorescencia: directa, indirecta e indirecta en piel separada.
La inmunofluorescencia directa (IFD) es un procedimiento de un solo
paso que se utiliza para demostrar el depósito de inmunoglobulinas,
complemento y fibrinógeno en la piel del paciente en estudio. La
inmunofluorescencia indirecta es una técnica de dos pasos que tiene
como objetivo la demostración de anticuerpos circulantes en el suero
del paciente dirigidos contra estructuras epidérmicas. La
inmunofluorescencia indirecta en piel separada es una técnica
que combina la inmunofluorescencia indirecta utilizando como substrato
una piel separada tras incubación con cloruro sódico. La
incubación con cloruro sódico provoca una separación
dermo-epidérmica al nivel de la lámina lúcida. La
realización de la inmunofluorescencia indirecta nos permite diferenciar
entre los anticuerpos dirigidos contra antígenos localizados en
la lámina lúcida (porción epidérmica, como
en el penfigoide ampolloso y herpes gestationis) y los dirigidos contra
la porción inferior de la membrana basal (epidermolisis ampollosa
adquirida, formas de penfigoide de mucosas y lupus eritematoso sistémico ampolloso).
|
Técnicas de inmunofluorescencia
|
|
La inmunofluorescencia directa (IFD) es un
procedimiento de un solo paso que se utiliza para demostrar el depósito
de inmunoglobulinas, complemento y fibrinógeno en la piel perilesional
del paciente. Para ello se obtiene una biopsia del paciente y se incuba
con anticuerpos dirigidos contra inmunoglobulinas humanas y marcados con
fluoresceina. Si estos se han fijado podrán ser visualizados mediante
un microscopio de inmunofluorescencia (Ultravioleta). |
La inmunofluorescencia indirecta
(IFI) es una técnica de dos pasos que tiene como objetivo la demostración
de anticuerpos circulantes en el suero del paciente dirigidos contra estructuras
epidérmicas. Para ello se incuba un sustrato (piel de animal) con
el suero del paciente en estudio y posteriormente se incuba con anticuerpos
marcados con fluoresceína dirigidos contra las inmunoglobulinas. |
|

|

|
5-Otras técnicas: Las
otras técnicas incluyen el
inmunobloting,
inmunoprecipitación, elisa, microscopia electrónica e inmunoelectromicroscopia,
son las técnicas que están aportando los conocimientos más
detallados de las enfermedades ampollosas autoinmunes, de tal manera que
gracias a estas técnicas estamos conociendo las características
de los diversos antígenos involucrados en las enfermedades
ampollosas lo que permite describir nuevas enfermedades ampollosas que
antes se engloban dentro de otras entidades. En la actualidad existen diversos
sistemas de ELISA comercializados para la detección de anticuerpos circulantes
contra diversos antígenos involucrados en las enfermedades ampollosas
autoinmunes (desmogleina 1 y 3, envoplaquina, BP180 y BP2320.
Entidades clínicas
De los datos obtenidos clínica y histológicamente se clasifican
las enfermedades ampollosas en intraepidérmicas y subepidérmicas
(tabla I)
|
TABLA I
CLASIFICACIÓN DE LAS ENFERMEDADES AMPOLLOSAS
|
|
Intraepidérmicas
|
Subepidérmicas
|
-
Pénfigo vulgar
-
Pénfigo foliáceo
-
Pénfigo IgA
-
Pénfigo paraneoplásico
|
-
Penfigoide ampolloso
-
Herpes gestationis
-
Penfigoide de mucosas (cicatricial)
-
Dermatosis IgA lineal infantil
-
Dermatosis IgA lineal del adulto
-
Epidermolisis ampollosa adquirida
-
Dermatitis herpetiforme
|
Las características histopatológicas
e inmunopatológicas del conjunto de las enfermedades ampollosas
están resumida en la
Tabla
II.
Enfermedades ampollosas intraepidérmicas: Pénfigos.
En este grupo se incluyen
las enfermedades ampollosas caracterizadas por la formación de ampollas
intraepidérmicas debidas a una pérdida de unión entre
las células intraepidérmicas (acantolisis) y caracterizadas
inmunopatológicamente por la presencia de autoanticuerpos circulantes
contra proteínas presentes en los
desmosomas
que conforman la unión intercelular. Existen varias formas de
pénfigos, en relación a la clínica y el tipo de anticuerpo
detectado. Las dos formas más conocidas de pénfigo, vulgar
y foliáceo, se deben a la presencia de anticuerpos contra los componentes
del desmosoma, concretamente la desmogleina 3 y 1. La clasificación
de las diferentes formas de pénfigo están resumidas en la
tabla III.
Pénfigo vulgar:
|
Tabla III
Clasificación de los pénfigos
|
Pénfigo
vulgar (desmogleina 3)
Pénfigo
vegetante tipo Newman
Pénfigo
vegetante tipo Hallopeau
Pénfigo
foliaceo (desmogleina 1)
|
Enfermedad ampollosa autoinmune
que afecta a la piel y mucosas caracterizada por la presencia de ampollas
intraepidérmicas suprabasales por acantolísis. Se caracteriza
por la presencia de anticuerpos anti sustancia intercelular de clase IgG,
dirigidos contra la desmogleina 3.
Clínica: Afecta
más frecuentemente a la cuarta o quinta década de la vida,
si bien se puede observar en cualquier edad. Puede observarse en cualquier
grupo racial, pero es más frecuente en pacientes con antepasados
judíos. Más del 25% de los casos se inician con manifestaciones
orales, que en ocasiones es la única manifestación. La lesión
clínica cutánea característica es la presencia
de una ampolla flácida, que se rompe fácilmente, dejando
amplias áreas erosivas. A nivel mucoso se observan ulceraciones
y erosiones superficiales amplias. Ocasionalmente pueden desarrollar lesiones
en otras localizaciones como cuero
cabelludo, lesiones digitales, ungueales, etc.
Microscopía óptica:
El pénfigo vulgar se caracteriza por una ampolla intraepidérmica
por acantolisis suprabasal, ocasionalmente puede existir un infiltrado
inflamatorio de predominio eosinófilo que puede llegar a afectar
a la epidermis dando la imagen de espongiosis eosinofílica. El test
de Tzanck es útil para observar la presencia de células acantolíticas
en el interior de la epidermis.
La inmunofluorescencia directa
(IFD), necesaria para confirmar el diagnóstico, muestra depósitos
de IgG en el espacio intercelular en el 90% de los enfermos, en un 30-50%
de los casos también se observan depósitos de C3. La inmunofluorescencia
indirecta (IFD) muestra anticuerpos circulantes en el suero de los pacientes
anti sustancia intercelular IgG en el 80-90% de los casos, el nivel de
estos anticuerpos se relaciona con la actividad de la enfermedad. Se ha
demostrado que estos autoanticuerpos están dirigidos contra desmogleina
3 (130Kd), que forma parte de la familia de las cadherinas presentes en
los desmosomas. En la actualidad existen métodos de ELISA para detectar
los niveles de estos autoanticuerpos. Los autoanticuerpos dirigidos contra
la desmogleina 3 presentes en los enfermos con pénfigo vulgar son
responsables de la acantolisis por interferencia directa en la función
de esta proteína en los mecanismos de unión intercelular. Existen cada vez más evidencias de que esta reacción autoinmune estaría
regulada por la acción de los linfocitos T
involucradas en la activación y diferenciación de las células B para
producir los anticuerpos patogénicos. Las manifestaciones clínicas del
pénfigo vulgar están en relación con la diferente distribución de las isoformas
de desmogleina. Los pacientes con penfigo vulgar que solo tienen anticuerpos
contra la desmogleina 3 tienden a tener lesiones localizadas en las mucosas. Los
pacientes con penfigo vulgar que tienen anticuerpos contra la desmogleina 1 y 3
tienen lesiones cutáneo mucosas generalizadas. Existen pacientes que
evolucionan de formas con predominio mucosa a formas mucocutaneas, o desde
formas de penfigo vulgar a formas a tipo penfigo foliáceo y más raramente en
sentido inverso. Este cambio en las manifestaciones clínicas generalmente
se relaciona con cambios en la reactividad del suero frente a las diferentes
isoformas de desmogleina. Una teroría para explicar este cambio se basa en la
extensión de epitopos, en la cual el daño tisular provocado por una respuesta
autoinmune o inflamatoria resultaría en la exposición de nuevos epitopos de la
misma u otra molécula produciende una respuesta autoinmune secundaria.
Curso: Antes de la instauración del tratamiento con corticoides,
el pénfigo vulgar era una enfermedad de pronóstico fatal
en la mayoría de los pacientes que fallecían por sepsis en
unos 5 años. Con la instauración de los corticoides, el pénfigo
vulgar tiene un pronóstico fatal en el 10% de los pacientes, en
general debido a las complicaciones del tratamiento.
Tratamiento: La utilización
de los corticoides en el tratamiento del pénfigo ha cambiado
el pronóstico de la enfermedad. Dado que las dosis de corticoides
utilizadas son altas (1-2 mg/kg/dia), en los casos en que se producen efectos
secundarios o no se consigue la remisión de la enfermedad, está
indicada la instauración de tratamiento adyuvante con inmunosupresores
(mofetil micofelonato, azatioprina, ciclofosfamida o clorambucil) y agentes
biológicos entre los que se encuentran la infusión de inmunoglobulinas y el
anticuerpo monoclonal anti CD20 rituximab.
Pénfigo vegetante:Variante rara de penfigo vulgar
caracterizada por el desarrollo de lesiones erosivas y pustulosas que afectan
predominantemente a los pliegues. Existen dos variantes, el tipo Neumann (forma
limitada) y el tipo Hallopeau (forma extensa), que están en relación con la
intensidad de las lesiones. Las manifestaciones
histopatológicas de ambas forma son semejantes a las del Pénfigo
vulgar observándose además acantosis y papilomatosis intensas
con formación de abscesos de eosinófilos intraepidérmicos.
Los hallazgos inmunopatológicos del pénfigo vegetante son
idénticos a los del pénfigo vulgar.
Pénfigo foliáceo:
Esta forma de pénfigo es una forma menos severa de la enfermedad en la cual la
ampolla se forma a un nivel más superficial de la epidermis. La enfermedad
afecta a personas ancianas y suele tener un curso lento, pero también puede
tener una evolución rápida instaurándose las lesiones descamativas de forma abrupta y dando la imagen de dermatitis exfoliativa.
Clínicamente las lesiones
del pénfigo foliáceo suelen ser ampollas superficiales que
se rompen con facilidad dejando áreas de la piel denudadas, localizándose
con mayor frecuencia en cara, cuello y tronco. No se acompaña de
lesiones mucosas.
Histológicamente el
pénfigo foliáceo se caracteriza por la presencia de una ampolla
subcórnea con un infiltrado inflamatorio en la dermis. La inmunofluorescencia
directa muestra depósitos de inmunorreactantes intercelulares a
nivel subcórneo. La inmunofluorescencia indirecta demuestra la presencia
de autoanticuerpos circulantes con un patrón semejante al del pénfigo
vulgar. Estos autoanticuerpos están dirigidos contra el dominio
extracelular de la desmogleina I (160Kd), produciéndose la ampolla al mismo
nivel que se produce en los casos de impétigo ampolloso y del síndrome de la
piel escaldada estafilocócica por la
toxina exfoliativa. La diferente distribución
en piel y mucosas en las diferentes capas epidérmicas
entre la desmogleina 1 y 3 ayudan a explicar la diferente expresión
clínica de ambas entidades.
Una variante de penfigo foliaceo
que se conoce como "Fogo Selvagen" ocurre de forma endémica en ciertas
áreas de Brasil. Otra variante está inducida por medicaciones,
especialmente la D-penicilamina y otra variante, pénfigo eritematoso
o síndrome de Senear-Usher, combina hallazgos clínico-patológicos
de pénfigo vulgar y de lupus eritematoso crónico cutáneo.
Pénfigo
herpetiforme: Es una variante de pénfigo foliaceo caracterizada por
la presencia de prurito, por la presencia de eosinófilos y neutrófilos
y por la respuesta al tratamiento con sulfonas. Combina hallazgos clínicos
de la dermatitis herpetiforme con hallazgos histológicos e inmunológicos
del pénfigo. Clínicamente se caracteriza por el desarrollo
de placas cutáneas eritematosas, con vesículas en la perifería
que con frecuencia siguen una distribución herpetiforme. Histológicamente
puede mostrar grados variables de acantolisis y de espongiosis eosinofílica.
La IFD muestra IgG intercelular superficial. Las técnicas de inmunoblot
demuestran que el autoantigeno característico de esta entidad es
la desmogleina 1.
Pénfigo
IgA: Es una variante de pénfigo caracterizada por la presencia
de anticuerpos circulantes y depósito intercelular de clase IgA.
Clínicamente desarrollan lesiones versículo-pustulosas, que
con frecuencia tienen una disposición anular afectando a axilas
e ingles, con escasa afectación de mucosas. Histológicamente
puede mostrarse con el desarrollo de pústulas que pueden tener una
disposición subcórnea o suprabasal, acompañados de
escasa acantolisis. La IFD muestra depósitos intercelulares de
IgA y en el 50% de pacientes la IFI muestra Ac circulantes de clase IgA.
Los estudios de inmunoblot sugieren que el autoantígeno del pénfigo
IgA sea la desmocolina 1, componente del desmosoma intercelular.
Pénfigo
paraneoplásico
Con anterioridad a la descripción
de esta forma de pénfigo, la asociación pénfigo-neoplasia
interna se consideraba fortuita. En la actualidad y tras diversos estudios
se ha establecido el penfigo paraneoplásico como una enfermedad
autoinmune que se observa en el curso de una neoplasia interna -benigna
o maligna (con mayor frecuencia linfoproliferativa)-en la cual se desarrollan
autoanticuerpos dirigidos contra antígenos involucrados en la unión
intercelular y dermo-epidérmica. Los estudios de inmunoblot e inmunoprecipitación
en el pénfigo paraneoplásico, han demostrado que los autoantígenos
responsables de esta entidad comprenden un grupo de proteínas que
conforman la placa desmosomica intracitoplásmica (Desmoplaquina
I, II, antígeno del penfigoide ampolloso, periplaquina) antígenos
desmosomales (desmogleina 1 y 3) y otros no determinados (ag 170kd) (Tabla
4).
Las lesiones clínicas se caracterizan por su gran polimorfismo, se inician en
forma de pápulas descamativas, pruriginosas, y posteriormente se desarrollan las ampollas.
La afección persistente de las mucosas y de las zonas acras es muy
común y las lesiones pueden conducir a un diagnóstico erróneo
de eritema polimorfo. El hallazgo clínico más constante es
la presencia de una estomatitis intratable, que generalmente es el primer
signo de la enfermedad y que tras tratamiento tiende a persistir. Esta
estomatitis consiste en el desarrollo de erosiones y ulceraciones que afectan
a toda la mucosa orofaríngea y que característicamente afectan
al borde bermellon de los labios. Las lesiones cutáneas muestran
mas variaciones entre pacientes y a lo largo del curso de la enfermedad,
consistiendo en ampollas flácidas, superficiales que se rompen con
facilidad. Las lesiones localizadas en piernas a menudo adoptan una morfología
clínica de lesiones a tipo eritema multiforme. Histológicamente
se observa una ampolla acantolítica con numerosos queratinocitos
necróticos así como una dermatitis de interfase. La IFD demuestra
depósitos de IgG y complemento en los espacios intercelulares de
la epidermis y depósitos de complemento granular /lineal en la membrana
basal. La IFI muestra depósitos intercelulares de clase IgG.
En todos los casos diagnosticados
hasta la actualidad el pénfigo paraneoplásico se ha asociado
con una neoplasia interna, en 2/3 de los casos el pénfigo
paraneoplásico se desarrolla en enfermos con neoplasia conocida
y en 1/3 la neoplasia se diagnostica tras la erupción cutánea.
Los tumores con los que más frecuencia se ha asociado son de origen
hematopoyético y por orden de frecuencia son linfomas no hodgkinianos,
leucemia, tumor de Castelman, timoma, macroglobulinemia y sarcomas. El
90% de los pacientes con pénfigo paraneoplásico fallecen debido a las complicaciones de la enfermedad de base o del tratamiento
inmunosupresor. El pénfigo paraneoplásico es la única
forma de pénfigo en que existe afectación visceral por el
proceso autoinmune que consiste en el desarrollo de acantolisis en el epitelio
bronquial. La afectación pulmonar con desarrollo de bronquiolitis
obliterante es la causa de muerte en un 30% de los pacientes.
Enfermedades ampollosas subepidérmicas
Penfigoides
El grupo de pengigoides lo constituyen
un conjunto de 8 enfermedades ampollosas autoinmunes caracterizadas por la
presencia de autoanticuerpos contra proteínas estructurales de la unión dermo-epidérmica
y clínicamente por el desarrollo de ampollas tensas y erosiones en la piel y
mucosas. El pronóstico y el tratamiento varía sustancialmente entre las
diferentes entidades y además de los criterios clínicos es necesario la
realización de técnicas de inmunofluorescencia directa en piel perilesional y
tests serológicos para establecer el diagnóstico ajustado.
|
 |
|
grupo de penfigoides
Datos clínicos relevantes y antígenos diana del grupo
de penfigoides |
|
|
Especificidad del autoanticuerpo (antígenos diana principales) |
Características clínicas |
|
Penfigoide ampolloso |
BP180
dominio NC16A
BP230 |
Ampollas
tensas y erosiones sin predominio de afectación mucosa |
|
Penfigoide de mucosas |
BP180,
laminina 332, BP230, α6β4 integrina,
laminina 311, colageno VII |
predominio
de afectación mucosa |
|
Herpes
gesttationis |
BP180
dominio NC16A
BP230 |
Lesiones
durante el embarazo o postparto |
|
Enfermedad IgA lineal |
LAD-1
BP230
(reactividad IgA) |
Ampollas
tensas y erosiones sin predominio de mucosas |
|
Epidermolisis bullosa adquirida |
Colágeno
tipo VII |
variante
inflamatoria y mecanoampollosa |
|
Pengigoide anti-lamininag1/anti-p200 |
Laminina
γ1(protína p200) |
Ampollas
tensas y erosiones sin predominio de mucosas |
|
Liquen
plano penfigoide |
BP180
dominio NC16A
BP230 |
Ampollas
tensas que no aparecen directamente en las lesiones de liquen plano |
|
Penfigoide
asociado a insuficiencia renal |
cadena α de colageno IV
antígenos no caracterizados
completamente |
|
Penfigoide ampolloso
Esta es la enfermedad ampollosa
autoinmune más frecuente, fue descrita inicialmente por W. Lever
en 1953. Consiste en una enfermedad ampollosa subepidérmica, de
curso crónico, de causa desconocida, caracterizada por la presencia
de autoanticuerpos dirigidos contra proteínas de los hemidesmosomas
conocidas como BP230 y BP180 (antígeno del penfigoide ampolloso
I y II).
Se caracteriza por afectar a personas mayores de 70 años.
Existen ciertos factores de riesgo para desarrolla un penfigoide ampolloso
que incluyen la presencia de enfermedades neurológica. Entre un tercio y la
mitad de pacientes con penfigoide ampolloso tienen enfermedades neurológicas
especialmente deterioro cognitivo importante, enfermedad de Parkinson,
Accidente cerebrovascular, epilepsia y esclerosis múltiples. Esta asociación
es llamativa debido a la expresión simultánea de los antígenos del
penfigoide ampolloso en el sistema nervioso central. Existen ciertos
factores desencadenantes que se han relacionado como traumatismos,
quemaduras o vacunas como la de la gripe. También se ha relacionado con ciertas medicaciones (espirinolactona, medicaciones dopaminérgicas y
psicolépticas y más recientemente con antidiabéticos de la familia de las
gliptinas (inhibidores de la dipeptidil peptidasa-4 o DPP-4). La incidencia de penfigoide ampolloso ha aumentado entre 2 y
5 veces en los últimos 10 años.
Clínicamente,
 los pacientes con penfigoide ampolloso desarrollan lesiones cutáneas y con menor frecuencia mucosas.
Las lesiones clínicas se caracterizan por ser máculas eritematosas
o urticariformes, pruriginosas que tras un período de tiempo variable
(semanas o meses) evolucionan hacia la formación de ampollas. Las
ampollas son grandes y tensas; cuando se rompen no se produce un despegamiento
de la epidermis en los bordes del área denudada y curan sin dejar
cicatriz. Las lesiones pueden distribuirse de un modo generalizado,
afectando preferentemente a la parte inferior del abdomen, las ingles,
las axilas y las superficies flexoras de los brazos y piernas, o bien localizadas,
preferentemente en miembros inferiores. El signo de Nikolsky es negativo.
Puede afectarse la mucosa oral en el 10-20% de los casos, por lo general
de forma leve, respetando el bermellón de los labios y nunca como
primera manifestación de la enfermedad. Existen diversas variantes
de la enfermedad entre las que destaca la forma de penfigoide ampolloso
de Brusting-Perry, caracterizada por la formación de placas eritematosas
y ampollosas, localizadas en cuero cabelludo, que curan dejando cicatriz.
los pacientes con penfigoide ampolloso desarrollan lesiones cutáneas y con menor frecuencia mucosas.
Las lesiones clínicas se caracterizan por ser máculas eritematosas
o urticariformes, pruriginosas que tras un período de tiempo variable
(semanas o meses) evolucionan hacia la formación de ampollas. Las
ampollas son grandes y tensas; cuando se rompen no se produce un despegamiento
de la epidermis en los bordes del área denudada y curan sin dejar
cicatriz. Las lesiones pueden distribuirse de un modo generalizado,
afectando preferentemente a la parte inferior del abdomen, las ingles,
las axilas y las superficies flexoras de los brazos y piernas, o bien localizadas,
preferentemente en miembros inferiores. El signo de Nikolsky es negativo.
Puede afectarse la mucosa oral en el 10-20% de los casos, por lo general
de forma leve, respetando el bermellón de los labios y nunca como
primera manifestación de la enfermedad. Existen diversas variantes
de la enfermedad entre las que destaca la forma de penfigoide ampolloso
de Brusting-Perry, caracterizada por la formación de placas eritematosas
y ampollosas, localizadas en cuero cabelludo, que curan dejando cicatriz.
El estudio histológico muestra una ampolla subepidérmica,
con infiltrado inflamatorio de predominio eosinófilo. La microscopia
electrónica muestra un hendidura a nivel de las células basales.
La
inmunofluorescencia directa muestra depósitos de IgG en
la UDE en ocasiones acompañándose de C3 y menos frecuentemente
de IgA, y IgM. La IFI muestra depósitos de anticuerpos circulantes
en el 70% de los pacientes, que en la técnica de piel separada se
observan dirigidos contra la porción epidérmica. La presencia de
autoanticuerpos circulantes contra el antígeno del penfigoide ampolloso
pueden ser detectados mediantes técninas de ELISA. Los niveles altos de los
autoanticuerpos pueden ser de utilidad para decidir reducir o suspender el
tratamiento ya que si persiste la elevación de autoanticuerpos, las
recidivas son frecuentes.

Existen amplias evidencias de que
el penfigoide ampolloso es resultado de reacciones de autoinmunidad celular y humoral contra el antígeno BP 180, concretamente
contra el dominio NC16A y otros epitopos del ectodominio BP180. La unión de
autoanticuerpos al Ag BP180, inicia una sería de eventos que incluyen la
liberación de interleucina 6 y 8 (IL-6 y IL-8), la activación del
complemento que induciría la qumiotaxis de células inflamatorias que
liberarían proteasas que serían las responsables de la separación dermo-epidérmica.
Existen evidencias realizadas en ratones transgenicos que replican las
características del penfigoide ampolloso y que han demostrado la existencia
de células T autoreactivas que podrían estar involucradas en la patogenia de
la enfermedad.
autoinmunidad celular y humoral contra el antígeno BP 180, concretamente
contra el dominio NC16A y otros epitopos del ectodominio BP180. La unión de
autoanticuerpos al Ag BP180, inicia una sería de eventos que incluyen la
liberación de interleucina 6 y 8 (IL-6 y IL-8), la activación del
complemento que induciría la qumiotaxis de células inflamatorias que
liberarían proteasas que serían las responsables de la separación dermo-epidérmica.
Existen evidencias realizadas en ratones transgenicos que replican las
características del penfigoide ampolloso y que han demostrado la existencia
de células T autoreactivas que podrían estar involucradas en la patogenia de
la enfermedad.
El diagnóstico de penfigoide
ampolloso se basa en la combinación de criterios clínicos, los estudios
histológicos, de inmunofluorescencia directa e indirecta. El tratamiento
consiste en la administración de corticoides, tópicos de alta potencia en
las formas moderadas y sistemicos a dosis de 0,5 mgr/kg/día de
prednisona.
 Herpes gestationis
Herpes gestationis
Enfermedad ampollosa subepidérmica
rara que se observa en el tercer trimestre del embarazo o en el período
postparto, con una frecuencia de 1 cada 50000 embarazos y menos frecuentemente
se asocia con tumores trofoblásticos. La enfermedad se origina en
presencia de tejido derivado del padre (feto). Se considera que un desajuste
entre el HLA del feto y de la madre desencadenaría una respuesta inmune
contra la piel materna. El herpes gestationis se considera
una variante del penfigoide ampolloso con el que comparte características,
clínicas, histológicos e inmunopatológicas.
Clínicamente se caracteriza
por una erupción
macular, urticariforme, que evoluciona hacia la formación de
vesículas, que inicialmente afecta al abdomen y para extenderse
posteriormente a otras áreas como extremidades, cara, y tórax.
La afectación de las mucosas es muy infrecuente. Histológicamente
se caracteriza por un intenso edema de la dermis papilar y con presencia
de espongiosis, así como por la presencia de focos de necrosis de
la capa basal. La inmunofluorescencia directa muestra el depósitos
de C3 en la unión dermo-epidérmica y menos frecuentemente
IgG. En la inmunofluorescencia indirecta sólo un 25% de los pacientes
muestra IgG anti membrana basal, sin embargo si se utiliza una técnica
de fijación del complemente se pueden detectar autoanticuerpos circulantes
en más del 90% de los pacientes. Estos anticuerpos están
dirigidos contra el antígeno del penfigoide ampolloso de 180 Kd.
Tratamiento: generalmente
se obtiene un buen control de la enfermedad con dosis bajas de prednisona
(20-40 mgr/día). El herpes gestationis es una enfermedad autolimitada
desapareciendo en las semanas o meses posteriores al parto, pudiendo recidivar
en embarazos posteriores.
Un 10% de los niños de madres con herpes gestationis pueden presentar
lesiones cutáneas transitorias. Menos del 5% de pacientes con
herpes gestationes evolucionan hacia penfigoide ampolloso.

 Penfigoide
Penfigoide de
mucosas
Penfigoide
Penfigoide de
mucosas
El penfigoide de
mucosas en un grupo de enfermedades ampollosas autoinmunes que afectan de
forma primaria a las mucosas especialmente oral y ocular y con menor
frecuencia a la piel. El término de penfigoide cicatricial se limita en la
actualidad a una variante menos frecuente en la que la afectación mucosa es
menos frecuente y las lesiones cutáneas curan con cicatrices. La enfermedad afecta especialmente a mujeres entre 50
y 60 años de edad. La afectación mucosa de forma crónica da lugar a la
formación de cicatrices que pueden acarrear complicaciones importantes con el
desarrollo de ceguera y asfixia. Clínicamente el penfigoide de mucosas
es una enfermedad crónica y progresiva que afecta predominantemente a la
mucosa oral
oral (85%) seguido de la
conjuntival (65%), piel (25-30%), mucosa nalsal (20-40%), región
anogenital (20%), faringe (20%), laringe (5-10%) y esófago (5-15%). Las
manifestaciones clínicas son muy variables desde pequelas lesiones
conjuntivales u orales a lesiones muy extensas con afectación mucosa y
esofágica. Las lesiones tienden a curar con cicatrización. La
afectación ocular suele iniciarse unilateralmente con síntomas menores como
sensación de quemazón o de cuerpo extraño pudiendo evolucionar hacia la
formación de cicatrices, sinblefaron, trichiasis, neovascularización y
ceguera. El penfigoide de mucosas se ha asociado con seis antígenos diana:
BP180 (˜ˍ75%), BP230 (25%), Laminina 332 (antes denominada laminina 5 o
epiligrina, 25%), ambas unidades de la α6β4 integrina y el colágeno tipo VII.El
diagnóstico del penfigoide de mucosas se basa en las caracerísticas
clínicas, en los hallazgos histológicos y en la biopsia de piel perilesiona
que mostrará hallagos indistinguibles del penfigoide ampolloso. Con técnicas
de inmunofluorescencia indirecta en piel separada con CLNa pueden observarse
hallazgos tanto epidérmicos como subepidérmicos. En los casos en que la
inmunofluorescencia indirecta en piel separada sea negativos o muestran
positividad en la porción dérmica debe realizarse una detección de
anticuerpos contra la laminina 332 ya que un 30% de pacientes que presentan
este anticuerpos presentan una
neoplasia
sólida asociada, de pulmón o gástrica, bien por la producción de
laminina por parte del tejido tumoral o por la exposición del antígeno en el
proceso inflamatorio o invasor del tumor son el origen de la producción de
los autoanticuerpos.
El tratamiento del penfigoide
cicatricial está en relación a la clínica y está
dirigido a evitar la formación de sinequias y cicatrices que pueden
desembocar en la ceguera. En los casos severos o de instauración
rápida es necesario realizar tratamientos con prednisona y ciclofosfamida.
En casos de enfermedad moderada puede iniciarse el tratamiento con sulfonas.
Los pacientes con afectación ocular deben seguir controles oftalmológicos
para evitar el desarrollo de secuelas.
Dermatosis ampollosa IgA lineal

Enfermedad ampollosa subepidérmica
caracterizada por el depósito de IgA lineal a nivel de la unión dermo-epidérmica.
Clínicamente se caracteriza por el desarrollo de ampollas polimorfas que en
ocasiones son similares a las del penfigoide ampolloso con ampollas tensas,
dispuestas en roseta, la presentación clínica en otras ocasiones es similar a la
de la dermatitis herpetiforme. Existen dos formas clínicas, la del adulto y la
infantil. La forma infantil, también conocida como enfermedad ampollosa crónica
de la infancia, suele afectar a niños de menos de 5 años, con lesiones
ampollosas que afectan frecuentemente a base de cuello y región genital.
La forma del adulto suele aparecer el la sexta década con lesiones ampollosas
localizadas preferentemente en tronco. Se han descrito formas inducidas por
medicaciones siendo la más frecuente la vancomicina. El estudio microscópico de
la IgA lineal muestra una ampolla subepidérmica con un denso infiltrado
inflamatorio de predominio polimorfonuclear neutrófilo. La IFD muestra los
depósitos de IgA lineales a nivel de la unión dermo-epidérmica. La IFI muestra
anticuerpos circulantes contra la unión y la IFI en piel separada muestra que
estos depósitos están dirigidos contra la porción epidérmica. La técnica de
inmunoblot ha identificado que el antígeno contra el que se dirigen los
autoanticuerpos es una proteína de 180 kDa, presumiblemente BP180, si bien se ha
determinado la presencia de varios antígenos involucrados. La
inmunoelectromicroscopía ha demostrado que la localización de los antígenos es
variable en los hemidesmosomas, lámina lúcida, lámina densa y sublámina densa.
Epidermolisis ampollosa adquirida (EBA)
(original)
Enfermedad ampollosa subepidérmica
con afectación de piel y mucosas caracterizada la presencia de autoanticuerpos
dirigidos contra el colágeno tipo VII, presente en las fibras de
anclaje de la unión dermo-epidérmica. Es una enfermedad infrecuente,
de causa desconocida, que afecta a la cuarta y quinta década, que
tiene una presentación clínica polimorfa. La forma clásica
se caracteriza por lesiones ampollosas de predilección en las zonas
acras, que aparecen tras traumatismos, que curan dejando cicatriz con formación
de quistes de milium. La histología muestra marcado edema
en dermis papilar y una ampolla subepidermica con grados variables de infiltrado
inflamatorio. La IFD muestra depósitos de IgG lineales en
la unión dermo-epidermica. La IFI muestra la presencia de autoanticuerpos
circulantes contra la unión dermo-epidermica y la IFI en piel separada
se observa que estos anticuerpos están dirigidos contra la porción
dérmica de la piel separada. La inmunoelectromicroscopía
demuestra que la localización de los depósitos de anticuerpos
en la EBA se produce a nivel de la sublámina densa y la técnica
de inmunoblot ha permitido caracterizar los antígenos de esta enfermedad
como dos proteínas que conforman el colágeno VII de 290 y
145 kDa. Un 20% de pacientes con epidermolisis bullosa
adquirida tienen una enfermedad inflamatoria intestinal asociada. La epidermolisis ampollosa adquirida responde mal a los tratamientos
habiéndose utilizado prednisona sola o en combinación con
otros inmunosupresores.
Penfigoide anti laminina γ1/Anti-p200
y liquen plano penfigoide
Son dos entidades raras. El anti-p200
simula un penfigoide ampollos, pero suele afectar a pacientes más jóvenes y 1/3
tienen asociada una psoriasis. El diagnóstico debe realizarse mediante
immunoblotting, por lo que probablemente está infradiagnosticado. El
liquen plano penfigoide siempre aparece asociado a liquen plano. El antígeno
diana es el BP180, afecta a pacientes más jóvenes de 40-50 años, desarrollando
lesiones principalmente en las piernas, de menor intensidad. El diagnóstico se
realiza mediane la observación de lesiones ampollosas en áreas diferentes de las
de liquen plano.
Dermatitis herpetiforme
La dermatitis herpetiforme
es una enfermedad vesículo-ampollosa, de curso crónico, caracterizada
por el desarrollo de vesículas intensamente pruriginosas agrupadas
afectando de forma simétrica a caras extensoras de extremidades
y por el depósito de IgA granular en dermis papilar asi como por
la asociación con una enteropatía por gluten generalmente
asintomática y con una respuesta beneficiosa a la instauración
de una dieta libre de gluten.
La etiología de la dermatitis herpetiforme no está
bien establecida, es probablemente inmunogenética, con una asociación
con aloantigenos HLA-DQ2.
Clínicamente
se caracteriza por la aparición de pápulas y vesículas
de distribución simétrica afectando a codos, rodillas, glúteos,
hombros y áreas sacras. El prurito acompañante a las lesiones
es en ocasiones la sintomatología más importante y puede
ser la inicial.
Histológicamente se
caracteriza por la formación de abscesos de polimorfonucleares en
las papilas dérmicas y formación de vesículas subepidérmicas.
La IFD muestra depósitos granulares de IgA en la dermis papilar,
estos hallazgos son imprescindibles para establecer el diagnóstico.
Recientemente se ha descrito que el autoantígeno involucrado en la dermatitis herpetiforme es la transglutaminasa
epidermica. Los pacientes con dermatitis herpetiforme y los pacientes
con enfermedad celíaca tienen anticuerpos circulantes contra la transglutaminasa y en los pacientes con DH se ha demostrado la presencia de
transglutaminasa asociada a IgA en las papilas dérmicas. El perfil de
anticuerpos antitransglutaminasa es diferente entre la enfermedad celíaca y la
DH, siendo más frecuentes los Ac antiTG2 en la enfermedad celíaca y los Ac
anti TG3 en la DH. Los anticuerpos antitransglutaminasa pueden medirse por ELISA. Los anticuerpos antiendomisio de clase
IgA, que se determinan por medio de inmunofluorescencia indirecta, tienen su
diana principal en la transglutaminasa hística. Los niveles de estos
anticuerpos son útiles para diagnosticar la dermatitis herpetiforme y la
enfermedad celíaca, pero sus niveles se negativizan si el paciente sigue dieta
exenta de gluten durante un tiempo prolongado.
Asociaciones sistémicas:
La mayoría de los pacientes afectos de dermatitis herpetiforme -sino
todos- están afectos de una enteropatía por gluten similar
a la que se observa en la celiaquía sin embargo, raramente presentan
sintomatología digestiva. Dentro de grupo infantil es más
frecuente observar la existencia de esteatorrea y alteración de
los test de absorción de D-Xilosa. En pacientes con celiaquia -sin
dermatitis herpetiforme-, existe un riesgo aumentado de desarrollar un
linfoma, está asociación también parece estar presente
en los enfermos con dermatitis herpetiforme.
Tratamiento: La dermatitis herpetiforme se puede tratar con sulfonas, con dieta exenta de gluten o
una combinación de ambas estrategias terapéuticas. La administración
de sulfonas a dosis de 50-100 mg día, produce un rápida resolución
de los síntomas, en un período muy corto de tiempo, sin embargo
no produce la desaparición de los depósitos de IgA en dermis
papilar ni de la enteropatía y se asocia con efectos secundarios
hematológicos. La instauración de una dieta libre de gluten
también produce una resolución de la sintomatología
pero puede requerir hasta 24 meses para ser efectiva y existen evidencias
de que reduce el riesgo de desarrollar linfoma. Alrededor del 10% de pacientes
una vez se ha obtenido control de la enfermedad pueden evolucionar hacia una
remisión sin precisar más tratamiento.
Tratamiento de las enfermedades ampollosas autoinmunes
El tratamiento de las enfermedades
ampollosas autoinmunes se basa en la utilización de agentes inmunosupresores
especialmente corticoides en dosis variables según la patología,
dependiendo de la severidad del cuadro se podrán utiilzar los corticoides
de diversas maneras, los enfermos con lesiones localizadas pueden tratarse
con corticoides topicos o intralesionales, los pacientes con enfermedad
moderada deben recibir tratamiento con corticoides orales y en aquellos
en que la enfermedad sea extensa y que no responda a los tratamientos orales
pude requerirse la utilización de corticoides endovenosos, tratamientos
de corticoides en pulsos
y tratamiento adyuvante con otros inmunosupresores. El resumen de los tratamientos
indicados para cada una de las entidades está resumido en la tabla
5.
casos per a discussió a classe de podologia 20240307 con asignación de alumnos
casos per a
discussió a classe
derm101quiz
www.uv.es/derma Dr. Víctor Alegre de
Miquel









