Linfomas cutáneos
Los linfomas cutáneos primarios
son aquellos procesos linfoproliferativos malignos –de linfocitos T ó
B- , cuya primera manifestación clínica es la presencia
de lesiones cutáneas sin existir enfermedad extracutánea
en el momento del diagnóstico pudiéndose observar afectación
ganglionar o visceral en el curso de la enfermedad. La piel es la segunda
localización (tras la localización gastrointestinal) en frecuencia de aparición de los linfomas primarios extranodales. A diferencia de los linfomas ganglionares no-Hodgkin que en su
mayoría son linfomas B, el 75% de los linfomas primarios cutáneos son linfomas T,
siendo en dos tercios de los casos micosis fungoide o síndrome de Sezary. La
incidencia de linfomas cutáneos T presenta un progresivo aumento siendo en la
actualidad de 6.4 casos por millón, con una relación ♂:♀ de 1.9. La incidencia
aumenta significativamente con la edad, siendo la edad media de diagnóstico los
50 años y es 4 veces más frecuente por encima de los 70 años.
Patogénesis de los linfomas
cutáneos
Existen varios mecanismos patogénicos
para el desarrollo de linfomas cutáneos que incluyen translocaciones
cromosómicas, alteraciones de los genes de supresión tumoral (bcl2),
alteraciones inmunológicas y factores externos como infecciones
víricas. La piel contiene unas características idóneas
para el desarrollo de linfomas. Se calcula que la piel normal contiene
aproximadamente 1 millón de células T por centímetro cúbico, consecuentemente la
piel es un órgano linfoide importante ya que contiene el doble de células T que
la sangre periférica. La piel, al igual que el ganglio linfático,
tiene varios compartimentos en las cuales se distribuyen los linfocitos
T y B. Por lo general los linfocitos T se distribuyen preferentemente en
la epidermis, dermis papilar, plexo vascular superficial, perianexialmente
y en la porción profunda del tejido graso subcutáneo (en
el ganglio se distribuyen en la región paracortical y médula). Los
linfocitos B se encuentran en la dermis media y profunda, plexo vascular
profundo y tejido graso (en el ganglio se distribuyen en el cortex y médula),
esta compartimentación está acompañada de un patrón
de secreción de citocinas característica. Esta distribución
de las subpoblaciones linfocitarias confiere un
patrón
histológico para las diferentes formas de linfomas cutáneos
primarios, así los linfomas cutáneos tipo micosis fungoide
presentan un patrón histológico epidermotropo mientras que
los linfomas B y T no micosis fungoide presentan un patrón difuso sin epidermotropismo
o perivascular.
Diagnostico de los linfomas
cutáneos primarios
La realización del diagnóstico
de linfoma cutáneo exige un abanico amplio de estudios clínicos
y patológicos para clasificar de forma correcta a los enfermos.
Estos estudios incluyen exámenes clínicos, radiológicos,
histológicos y estudios moleculares. Los pacientes con sospecha
de linfoma cutáneo primario deben someterse a un examen clínico
completo incluyendo estudios radiológicos tóraco-abdominales
(Rx tórax, TAC toraco-abdominal) y examen de médula ósea, todos ellos en
relación con el diagnóstico de
sospecha
. Los
estudios histológicos deben incluir el estudio de hematoxilina eosina,
PAS y Giemsa. Deben realizarse asimismo estudios inmunohistoquimicos con
anticuerpos contra antígenos
característicos de las diferentes subpoblaciones celulares de los
linfocitos, macrófagos, células interdigitantes y dendríticas.
Los análisis de reordenamiento genético mediante técnicas
de Southern blot y de PCR (PCR/DGGE) de los genes del receptor de células
T (TCR) y de inmunoglobulinas son técnicas útiles en
el estudio de los linfomas cutáneos dado que pueden aporta datos
sobre la presencia de clonas
de linfocitos.
El diagnóstico final va a depender de la integración de los datos clínicos e
histológicos, teniendo en cuenta la evolución de la enfermedad.
Clasificación de los
linfomas cutáneos
Existen diversas formas de clasificar
los linfomas, recientemente se ha ha llegado a un consenso de clasificación que
agrupa la clasificación de la OMS y EORTC (blood
2005) que
sustituye a las previas e independientes de la OMS
y EORTC
.
Linfomas cutáneos
de células T
Micosis Fungoides
Es la forma más frecuente
de linfoma cutáneo primario, se trata de un linfoma cutáneo de células T,
frecuentemente epidermotropo, que se caracteriza por una proliferación de
linfocitos T de pequeño o mediano calibre con núcleo cerebriforme. Fue inicialmente descrita en 1806 por Alibert
recibiendo su nombre por el aspecto fungoide de las lesiones tumorales
cutáneas. Tiene una incidencia de 0,29 casos /100.000 habitantes
y año, con un pico de edad entre los 55 y 60 años. Las manifestaciones
clínicas son muy variables desde lesiones únicas hasta la eritrodermia.
La
presentación más clásica es el desarrollo de máculas y placas, bien delimitadas, eritematosas, con descamación variable con predilección por localizarse en las
nalgas y otras áreas protegidas de la luz solar. Las lesiones pueden progresar hacia la formación de
placas y tumores. En muchas ocasiones las lesiones son poiquilodérmicas con
presencia de atrófia epidérmica, telangiectasias y áreas moteadas de hiper e
hipopigmentación.
Formas
clínicas de Micosis fungoide: La micosis fungoide tiene un curso
clínico indolente con una progresión lenta a lo largo de los años y a veces
décadas en que las lesiones pasa de un estadio macular o eczematoso a el
desarrollo de placas y tumores.
|
Fases clínicas de
la micosis fungoide |
|
Mácula |
Lesión seca, elevación de tamaño variable sin induración
ni elevación significativo con cambios pigmentarios , escamas y costras |
|
Placa |
Lesión indurada y elevada de tamaño variable con cambios
pigmentarios |
|
Tumor |
Lesión nodular, sólida de >1 cm o ulcerada con evidencias
de crecimiento exofitico |
|
Eritrodermia |
Confluencia de lesiones eritematosas cubriendo >80% de la
superficie cutánea |
|
MF eritrodermica |
Eritrodermia sin evidencia clínica de afectación
hematológica |
|
Síndrome de Sezary |
Eritrodermia con afectación hematológica evidente |
- Estadio Macular o eczematoso:
Se caracteriza por maculas eritematosas de tamaño variable, cubiertas
de escamas finas y pequeñas, generalmente muy pruriginosas. Existe
una forma denominada “digitata” en la cual las lesiones adoptan la morfología
de la huella digital.
- Estadio en placas: se caracteriza
por lesiones cutáneas sobreelevadas, infiltradas, eritemato-descamativas,
con frecuencia asociadas en periferia se observan lesiones maculares o
eczematosas.
- Estadio tumoral: consiste en
la presencia de lesiones cutáneas de mayor tamaño, eritematosas,
azuladas, infiltradas, con frecuencia ulceradas, asociadas a otras
lesiones clínicas (maculares y en placas). Su presencia indica un
peor pronóstico
Estudios de laboratorio
Histología:
Los
hallazgos histológicos de la micosis fungoide consisten en la presencia de un
infiltrado linfoide en dermis papilar con distribuido en banda y/o afectando a
la unión dermo-epidérmica, existiendo presencia de células linfoides en la
epidermis sin acompañarse de espongiosis. El infiltrado linfoide está
constituido por células de núcleo hipercromático, irregular y convoluto (cerebriforme).
Cuando se observa dentro de la epidermis las células con frecuencia están
rodeadas de un halo claro y suelen ser de mayor tamaño que las observadas en la
dermis. Ocasionalmente pueden observarse colecciones intraepidermicas de células
linfoides que se conocen como
microabscesos de Pautrier y son
característicos de los linfomas cutáneos de células T y reflejan la interacción
entre las células de Langerhans y los linfocitos T neoplásicos. En los linfomas cutáneos de células T, las
células de linfoma tienen predilección por la epidermis y eso es debido a la
interacción-mediada por el patrón de secreción de citocinas y por la presencia
de los diferentes receptores en la superficie celular-entre las células del
linfoma y tanto con las células de los endotelios capilares de la dermis así
como con las células de Langerhans alrededor de las cuales las células del
linfoma se agrupan formando los microabscesos que son visibles histológicamente.
Inmunofenotipo: El
inmunofenotipo de la micosis fungoide muestra que se trata de un linfoma T
periférico constituido por células T (CD3+), helper o cooperadoras (CD4+,CD8-
(ocasionalmente pueden ser CD4-CD8+)), de
memoria (CD45Ro+) que pertenecen al tejido linfoide asociado a la piel (SALT,
CLA+) y que además suelen perder la expresión de CD7.
Genética: con
la técnica de PCR/DGGE es posible demostrar la clonalidad en hasta el 90% de los
casos de linfoma cutáneo de células T
Pronóstico y estadiaje de la micosis fungoide: La extensión de la enfermedad puede ser variable
así como la afectación visceral, siendo útil la clasificación
TNM
para el estadiaje de los pacientes con micosis fungoide (y
TNM para linfomas
cutáneos no micosis fungoide). De cara a comprender la
clínica, evolución y planificar el tratamiento de la micosis fungoide es
necesario entender el
origen y el
comportamiento de las células T en esta enfermedad.
La micosis fungoide
si bien es un linfoma presenta en los
estadios iniciales tiene un buen pronóstico y la evolución
a estadios en placa o tumoral es impredecible. Los estadios cutáneos
iniciales (T1) tienen una supervivencia superior a los 30 años, falleciendo
los pacientes por otras causas. Cuando la afectación es únicamente
cutánea, se obtienen buenas respuestas con tratamientos que pueden
incluir la aplicación de corticoides tópicos, fototerapia
con UVB o PUVA de forma aislada o asociada a interferón alfa 2a. Dado que los
linfocitos T neoplásicos son radiosensibles, la utilización de la irradiación
corporal total de toda la superficie cutánea es muy útil en las fases iniciales
de la enfermedad con respuestas superiores al 90%. En pacientes con enfermedad
avanzada es necesario realizar una aproximación terapéutica multidisciplinar con
tratamientos dirigidos a la piel, tratamientos modificadores de la respuesta
biológica y ocasionalmente radioterapia.
|
Opciones
terapéuticas en los linfoma cutáneos de células T |
|
Tratamiento local |
Tratamiento sistémico |
|
Corticoides
topicos de alta potencia
PUVA/UVB/helioterapia
Bexaroteno
Mostaza
nitrogenada
Radioterapia |
PUVA + Interferon alfa2b
Metotrexate
Clorambucil/ciclofosfamida
Bexaroteno
Poliquimioterapia |
|
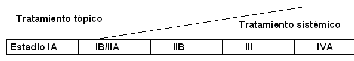 |
Transformación a linfoma
cutáneo de célula grande: En las fases avanzadas de la MF,
los pacientes pueden desarrollar lesiones tumorales con presencia de células
grandes, la transformación a linfoma de célula grande se
considera cuando las células grandes suponen más del 25%
de las presentes en el infiltrado y su presencia implica un más
pronóstico
Síndrome
de Sezary: Es una variante rara y agresiva de linfoma cutáneo de células T,
caracterizada por afectación cutánea y leucémica. Los estudios moleculares han
demostrado que las células tumorales en el síndrome de Sezary derivan de las
células T de memoria centrales, a diferencia de las células neoplásicas de la
micosis fungoide que derivan de las células T de memoria efectoras.
Clínicamente se caracteriza por eritrodermia
muy pruriginosa asociada a linfadenopatía generalizada y la
presencia en piel, ganglios y sangre periférica de linfocitos T
atípicos (Células
de Sezary). Afecta a personas ancianas
de ambos sexos y se caracteriza por el desarrollo de una eritrodermia generalizada
con descamación fina y muy pruriginosa, que puede cursar con hiperpigmentación
cutánea. Es frecuente la asociación a hiperqueratosis palmo-plantar
y onicodistrofia. La presencia de linfadenopatía generalizada es
constante y el estudio histológico de los ganglios linfáticos
suele ser positivo y específico. Los hallazgos histológicos
del síndrome de Sezary son similares a los de la MF, con presencia
de un infiltrado denso, en banda, en dermis papilar, que por lo general
muestra menor tendencia al epidermotropismo. El estudio inmunofenotípico
muestra la presencia de linfocitos T, con predominio de T cooperadores
(CD3+ CD4+, CD8-). La mayoría de los casos muestra reagrupación
clonal del receptor de linfocitos T (TCR) en la sangre periférica, que se
considera un criterio diagnóstico del síndrome de Sezary. El diagnóstico de Síndrome
de Sezary se basa en la presencia de una o más de los siguientes hallazgos:
El pronóstico es generalmente malo con
una supervivencia media de entre 2 y 4 años. El tratamiento es por lo general
poco satisfactorio, se obtienen resultados con fotoferesis extracorporea,
alemtuzumab y quimioterapia
Linfomas cutáneos de células T, distintos de la micosis fungoide
Linfomas cutáneos
primarios CD30+:
después
de la micosis fungoide es el grupo de linfomas cutáneos primarios más frecuente
e incluye la papulosis linfomatoide y el linfoma cutáneo T de célula grande
CD30+ .
Papulosis
Linfomatoide: Síndrome caracterizado
lesiones cutáneas pápulo-nodulares de pequeño tamaño, recidivantes, con
tendencia a la ulceración y curación espontánea dejando cicatrices atróficas y
que se caracteriza por hallazgos histológicos similares a los encontrados en los
linfomas cutáneos de células T (CD30+), este síndrome se clasifica dentro de los
linfomas T. Afecta principalmente a adultos jóvenes y se caracteriza por
una erupción generalizada de pápulas o nódulos de
pequeño tamaño que afectan especialmente a tronco y extremidades,
las lesiones son eritematosas, parduzcas y con tendencia a la curación
espontánea (las lesiones tienden a durar entre 3 y 6 semanas)
y con tendencia a cursar en brotes, con una evolución prologada
(de entre 3 meses y 40 años), siendo frecuente la observación
de lesiones en diferentes estadios de evolución.
Histológicamente se caracteriza por un infiltrado dérmico
de linfocitos T cooperadores habiéndose descrito diversas variedades histológicas:
el tipo A (tipo histiocítico) caracterizado por la presencia de
células grandes y atípicas entremezcladas con los linfocitos
pequeños, histiocitos y eosinófilos; el tipo B (tipo micosis-fungoide,
tipo mas infrecuente)
con hallazgos similares a la micosis fungoide con presencia de linfocitos
atípicos y cerebriformes, el tipo C (tipo similar al linfoma anaplásico
de célula grande), caracterizado por un denso infiltrado nodular
de células grandes atípicas entremezcladas con linfocitos
pequeños neutrófilos y eosinófilos, con hallazgos
indistinguibles de los linfomas anaplásicos de célula grande y el
tipo D caracterizado por un infiltrado epidermotropo de células pequeñas y
medianas atípicas CD8+ y CD30+. Inmunofenotípicamente el infiltrado es CD3+, CD4+, CD8-, las células
grandes presentes en los tipos A y C son CD30+. Un 60-70% muestran reordenamiento
clonal del TCR. La papulosis linfomatoide suele presentar un pronóstico
excelente reduciéndose la frecuencia de síntomas con la administración
de metotrexate a dosis bajas, PUVA (solo o con IFNα2a) o corticoides.
Linfoma
cutáneo T de célula grande (CD30+) :
Se trata de
un linfoma que se caracteriza por el desarrollo de lesiones nodulares generalmente
solitarias y caracterizadas histológicamente por un denso
infiltrado de linfocitos de gran tamaño, anaplásicos y que
expresan en un alto porcentaje el antígeno CD30+. Afecta
a adultos que desarrollan lesiones nodulares, ulceradas, en general solitarias,
rodeadas de un halo eritematoso. Histológicamente se caracterizan
por un denso infiltrado nodular o difuso compuesto por células grandes
atípicas de núcleo irregular con nucleolo prominente semejando
a las células de Reed-Sternberg, observándose asimismo células
grandes pleomórficas. La mayor parte de las células del infiltrado
tienen fenotipo de células T cooperadoras (CD3+, CD4+, CD8-) y son
CD30+. La mayoría de los casos muestran reordenamiento clonal del
TCR. Las lesiones aisladas suelen responder bien a la radioterapia
o a la extirpación quirúrgica.
Linfomas cutáneos de células B
Los linfomas B primarios cutáneos son mucho menos frecuentes que los linfomas T,
representando entre un 20-25% de todos los linfomas primarios cutáneos. Existen
tres formas principales: linfoma primario cutáneo de la zona marginal, linfoma
primario cutáneo del centro folicular y linfoma primario difuso de célula grande
tipo piernas.
|
Formas de linfoma cutáneo primario B |
|
Tipo (Clasificación OMS 2008) |
linfoma primario cutáneo de la zona
marginal |
Linfoma primario cutáneo del centro
folicular |
Linfoma primario cutáneo difuso de
célula grande B, tipo piernas |
|
Características clínicas |
Pápulas, placas y nódulos,
solitarios o múltiples, localizados preferentemente en extemidades.
Ocasionalmente relacionado con
infección por Borrelia burgdorferi
Las recidivas cutáneas son
frecuentes.
Raramente existe afectación
extracutánea |
Tumores solitarios o agrupados
localizados en cabeza o tronco
20% presentan recidivas cutáneas
5-10% presentan diseminación
extracutánea |
Tumores solitarios o múltiples,
localizados preferentemente en las piernas y solo raramente en otras
localizaciones
Recidivas frecuentes y afectación
extracutánea |
|
Histología |
Infiltrado parcheado o difuso
compuesto por células B pequeñas, incluyendo células de la zona marginal
(tipo centrocitos), células linfoplasmocitoides y células plasmáticas |
Infiltrado difuso, difuso y
folicular o folicular, compuesto por células del centro folicular,
generalimente una mezcla de centrocitos y centroblastos |
Infiltrado difuso con un predominio
de áreas de inmunoblastos y centroblastos |
|
Inmunofenotipo |
Inmunoglobulinas de citoplasma
monotipicas, CD79a+, Bcl-2+, CD5-neg, ciclina D1-neg, Bcl-6-neg, CD10-neg,
MUM-1+ (en células plasmáticas). |
Inmunoglobulina de superficie
monotipica o ausente, CD20+, CD79a+, Bcl-6+, Bcl-2+, MUM-1-neg, CD10+/-,
FOXP1 -(+/-) |
Inm.unoglobulina de superficie
o citoplasmática monotipica, FOXP1+, CD79a+, Bcl-6+(-), CD10-neg, Bcl-2+, MUM-1+,
|
|
Pronóstico |
supervivencia a los 5 años> 95% |
Supervivencia a los 5 años: 95% |
Supervivencia a los 5 años: 50% |
Linfoma cutáneo de células B de la
zona marginal primario cutáneo: Es una forma de linfoma indolente
formado por células B pequeñas, como células de la zona marginal, células linfo-plasmocitoides
y células plasmáticas. Clínicamente se caracteriza por el desarrollo de pápulas,
placas o nódulos, de color rojo o violáceo, localizados preferentemente en
tronco, frecuentemente multifocales. Las lesiones tienen tendencia a la recidiva
pero su localización extracutánea es muy infrecuente. En una minoría de casos
europeos se ha demostrado una asociación con la infección por Borrelia
burgdorferi. Histológicamente se caracteriza por un infiltrado nodular o
difuso, sin afectación de la epidermis, constituido por linfcitos B pequeños,
células B de la zona marginal, células linfoplasmocitoides y células
plasmáticas. Inmunofenotípicamente son células CD20+, bcl-2+ y muestran
reordenación clonal para las cadenas pesadas de inmunoglobulinas. El pronóstico
en estos casos es excelente con una supervivencia a los 5 años superior al 95%. El
tratamiento de las lesiones localizadas puede realizarse mediante radioterapia o cirugia.
Linfoma
centro folicular: Representa la proliferación
neoplásica de las células del centro germinal, con
presencia de centrocitos (células del centro folicular pequeñas
y grandes de núcleo hendido) y centroblastos (células del
centro folicular grandes y de nucleolo prominente) limitado a la piel.
Clínicamente se caracteriza por el desarrollo de pápulas,
placas o tumores, aislados o en grupos rodeados de máculas eritematosas,
afectando preferentemente varones con una edad media de 60 años, estando
localizado en frente, cuero cabelludo y espalda, raramente
presentan ulceración. Las lesiones tienen un comportamiento progresivo
lento, aumentando de tamaño con los años, pero la diseminación extracutánea es muy rara. Histológicamente se caracterizan por un infiltrado dérmico
y subcutáneo nodular o difuso, sin afectación de la epidermis,
que solo raramente se dispone con un patrón folicular. No suele
existir afectación de la epidermis, Las células del infiltrado
adoptan la morfología de centroblastos y centrocitos. Inmunofenotipicamente
son linfocitos B (Inmunoglobulinas de superficie +, CD20+(antígeno
pan B), CD10+ y bcl-6+) y la detección de la proteína BCL-2
es negativa lo cual los diferencia de los linfomas nodales. La mayoría
de los casos presenta reordenamiento clonal del gen de las inmunoglobulinas.
El tratamiento de elección es la radioterapia, siendo frecuentes
las recidivas.
Pseudolinfomas cutáneos
Los pseudolinfomas cutáneos
son enfermedades inflamatorias de la piel que semejan clínica y/o
histológicamente linfomas. Se dividen en pseudolinfomas de células
B y T en relación al patrón histológico e inmunopatológico
que presentan.
Pseudolinfomas T
- Reticuloide actínico:
esta entidad es una reacción inflamatoria crónica que es
resultado de una dermatitis fotoalérgica persistente. Suele
afecta a varones mayores. Clínicamente se caracteriza por el desarrollo
de pápulas y placas eritemato-violáceas, liquenoides que
afectan a zonas expuestas y que en ocasiones desarrollan eritrodermia muy
pruriginosa, acompañada de alopecia y linfadenopatía generalizada
pudiéndose observar la presencia de células de Sezary en
sangre periférica. El estudio histológico muestra la existencia
de cambios liquenoides en la epidermis y con un infiltrado de linfocitos
atípicos que focalmente pueden mostrar epidermotropismo. El inmunofenotipo
muestra fenotipo de células T supresoras. El pronostico es bueno
siendo muy importante evitar la luz solar para mejorar los síntomas.
-
Reacciones medicamentosas linfomatoides a tipo micosis fungoide ciertos medicamentos –antidepresivos
y antihistamínicos. En ocasiones producen una reacción cutánea
medicamentosa con hallazgos característicos de la micosis fungoide
Pseudolinfomas B
-
Linfadenosis cutánea
benigna (linfocitoma cutis): es el ejemplo más característico
de pseudolinfoma B, puede estar inducido por diferentes estímulos
incluyendo picaduras de insectos, vacunas, tratamientos de hiposensibilización,
tatuajes, etc. Se caracteriza clínicamente por el desarrollo de
lesiones cutáneas generalmente únicas, papulares o
nodulares, eritematosas e infiltradas. Afectan especialmente a áreas
como la cara (mejilla, nariz, lóbulos auriculares), tronco (pezones),
escroto y extremidades. El estudio histológico muestra un infiltrado
nodular denso, polimorfo con presencia de linfocitos que ocasionalmente
forman nódulos linfoides con centros germinales, células
plasmáticas y eosinofilos. Inmunofenotípicamente existe un
predominio de células B –CD20+-. Las lesiones pueden resolverse
espontáneamente tras largos períodos o tras tratamiento
que puede incluir la inyección intralesional de esteroides y/o radioterapia.
Mastocitosis
Las mastocitosis son un grupo
heterogéneo de entidades caracterizadas por el aumento del número
y acumulación de mastocitos en uno o más órganos.
La piel es el órgano que se afecta con mayor frecuencia, su afectación
puede ser la única manifestación (mastocitosis cutánea)
o estar afecta en el conjunto de una mastocitosis sistémica. No
se conocen los mecanismos patogénicos de las mastocitosis. El hallazgo
de mutaciones en la molécula c-kit
en los casos de mastocitosis sistémicas sugiere la posibilidad de
que las mastocitosis (al menos las sistémicas) sean debidas a una
proliferación clonal de los mastocitos. El aumento en el número
de mastocitos y la liberación de sus mediadores (histamina, citocinas,
leucotrienos) son responsables de los efectos locales y sistémicos
de estas entidades.
Las mastocitosis se dividen
en varias categorías en relación si son indolentes (cutáneas
o sistemicas indolentes), asociadas a hemopatías, agresivas o la
forma de leucemia de mastocitos (tabla
I). Las formas cutáneas y sistémicas indolentes tiene
por lo general un buen pronóstico, en las formas asociadas a hemopatías
el pronóstico depende de la hemopatía asociada y en la forma
de leucemia a mastocitos el pronóstico es infausto.
Mastocitosis
cutáneas: La mastocitosis cutánea es la forma más
frecuente de la enfermedad, afecta especialmente a niños (con mayor
frecuencia menores de 2 años) y tienen por lo general un buen pronóstico. Este
grupo de mastocitosis se caracteriza por el desarrollo de lesiones cutáneas
típicas, con signo
de Darier positivo y presencia en las biopsias cutáneas de infiltrados
de mastocitos y en ausencia de criterios de afectación sistémica
así como ausencia de mutaciones de c-kit. Existen 4
formas clínicas de mastocitosis cutánea:
- Urticaria pigmentosa:
es la forma más frecuente (85% de casos) de mastocitosis cutánea.
Se caracteriza por el desarrollo de máculas y pápulas parduzcas
o amarillentas, en número variable -desde unas pocas a cientos-
distribuidas por todo el tegumento. Las lesiones clínicas presentan
un signo de Darier positivo. Las formas de urticaria pigmentosa que aparecen
en la infancia tienen a desaparecer espontáneamente y no precisan
evaluación hematológica. Las formas de aparición en
la edad adulta presentan una alta asociación con afectación
sistémica.
- Mastocitoma: constituye
un 15% de las formas de mastocitosis cutáneas. Se caracteriza por
la presencia de una placa o nódulo amarillento, generalmente
solitario, que al rascado presenta un signo de Darier muy positivo que pude dar
lugar a la formación de ampollas
- Mastocitosis cutánea
difusa: es una forma rara de mastocitosis en la que existe infiltración
de toda la piel con engrosamiento cutáneo difuso más llamativo
a nivel de los pliegues. La afectación sistémica en estos
casos es más frecuente.
- Telangiectasia macular eruptiva perstans: es una forma rara de mastocitosis cutánea que se observa
en adultos y se caracteriza por el desarrollo de máculas pigmentadas
con presencia de telangiectasias, el signo de Darier en estos casos puede
ser negativo.
Histología: El estudio
histológico muestra la presencia de un aumento en el número
de mastocitos que se disponen en dermis papilar y media, con distribución
preferentemente perivascular y que se tiñen metacromáticamente
con el azul de toluidina o la tinción de Giemsa.
Mastocitosis sistémica:
Las
mastocitosis sistémicas se observan especialmente en adultos en
los que se puede evidenciar histológicamente la afectación
de médula ósea y de otros órganos, así como por la presencia de marcadores
serológicos y citológicos de la enfermedad (demostración
de la mutación c-kit, elevación de los niveles de triptasa
serica (>20 ng/ml). Pueden tener diversos síntomas clínicos
que podrán depender en relación a si son debidos a la enfermedad
sistémica (síntomas constitucionales: pérdida de peso,
malestar, cansancio), a la infiltración tisular por los mastocitos
(piel, médula ósea, pulmón, tracto digestivo) o a
la liberación de mediadores (síncope, shock, diarrea y dolor
abdominal, ulcus péptico, dolor óseo, cefalea severa, flushing
recurrente).El desarrollo de una mastocitosis en la edad adulta confiere
pues un elevado riesgo de afectación sistémica (75%) y hasta
un 60% de los pacientes con mastocitosis sistemica desarrollaran neoplasias
hematológicas.
casos para discusión
www.uv.es/derma Dr. Víctor Alegre de
Miquel

{kind=link}