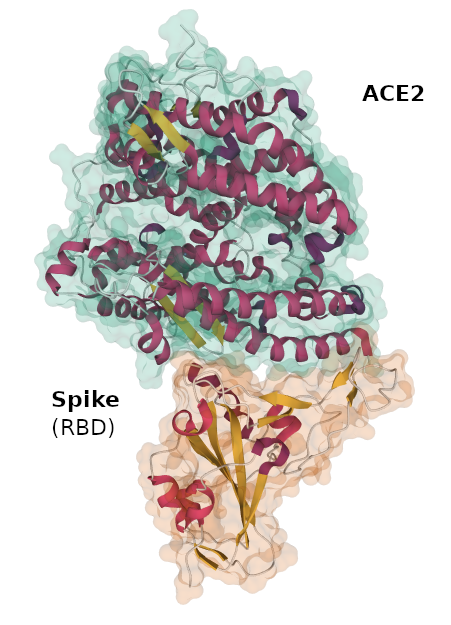
Universitat de València - Máster en Aproximaciones Moleculares en Ciencias de la Salud
Tecnologías de la Medicina Molecular
Modelización y Análisis de Estructuras Macromoleculares
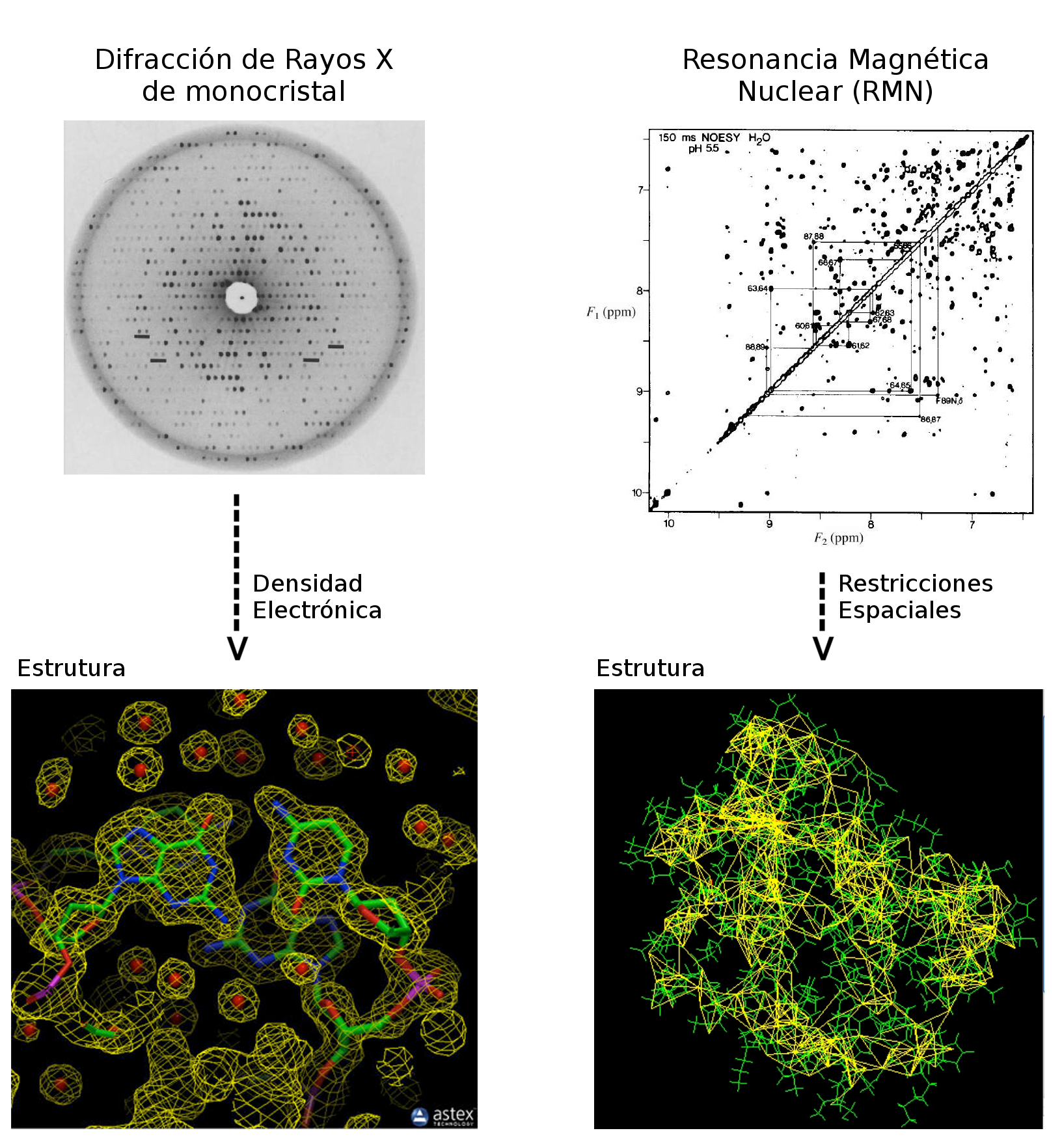
Determinación Experimental de Estructuras de Proteínas
En la actualidad se conocen con detalle atómico mas de 2000000 estructuras tridimensionales de macromoléculas biológicas. Cada una de ellas ha sido determinada experimentalmente, utilizando distintos métodos, principalmente Difracción de Rayos-X (~90% de los casos), Resonancia magnética Nuclear (RMN) (~9% de los casos), y en menor medida, Microscopía Electrónica y otras técnicas de difracción.
Toda esta enorme información estructural está disponible de forma libre para la enseñanza, la investigación la sanidad o la industria. Para ello se utiliza un formato especial (formato pdb), en el cual se escribe la estructura de cada proteína en un fichero de texto y se le asigna un código propio de identificación (pdb-ID). Todos los ficheros de estructuras se depositan y organizan en un archivo único accesible sin restricciones a través de Internet.
El archivo único de estructuras 3D de macromoléculas es gestionado a nivel Mundial a través del Consorcio Worldwide Protein Data Bank (wwPDB), formado por cuatro miembros: dos en USA (RCSB PDB y BMRB), uno en Europa (PDBe) y uno en Japón (PDBj). Cada uno de ellos organiza el deposito de nuevas estructuras, a la vez que mantiene una copia del archivo de estructuras completo, a través de las bases de datos RCSB PDB, PDBe y PDBj.
Las bases de datos facilitan el acceso a la información estructural a través de herramientas de búsqueda. Además, proporcionan información estadística sobre el contenido del archivo e información bibliográfica sobre cada registro, y complementan la información estructural depositada por los autores con una amplia gama de nuevos datos obtenidos a partir del análisis de las estructuras, tales como:
Las estructuras de macromoléculas biológicas corresponden a una escala nanométrica, por lo que no pueden ser observadas directamente con resolución atómica. Por ello, para su estudio se llevan a cabo representaciones gráficas a partir de los ficheros de coordenadas atómicas experimentales y con ayuda de programas informáticos. Dichas representaciones computacionales son modelos moleculares tridimensionales que pueden ser rotados, anotados y re-escalados y que en muchos casos permiten el cálculo de propiedades moleculares.
Existe una gran variedad de programas informáticos adecualdos para llevar a cabo representaciones gráficas de macromoléculas biológicas. En general, pueden clasificarse en:
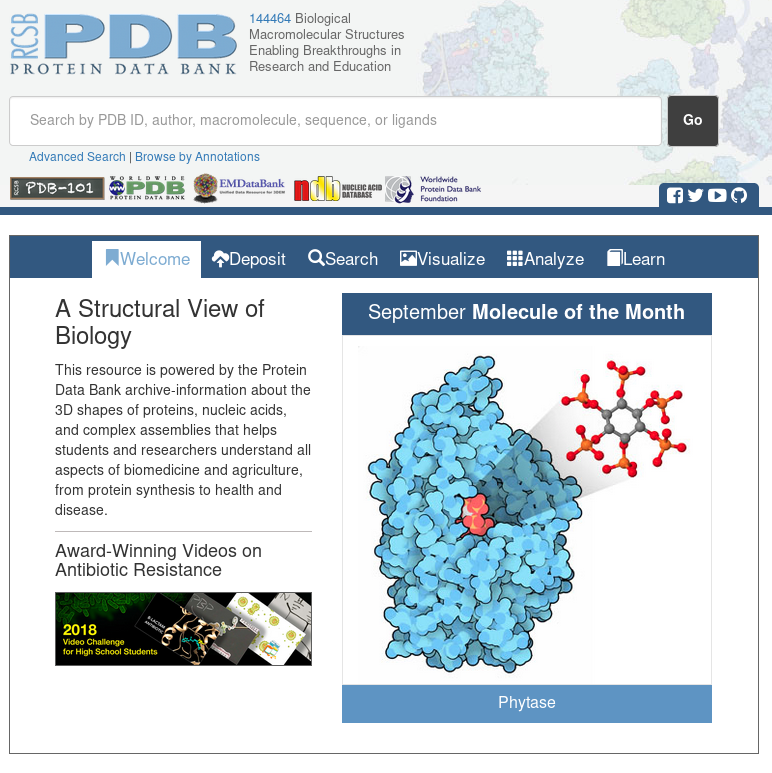
Protein Data Bank, en www.rcsb.org
El consorcio internacional wwPDB mantiene un repositorio único de estructuras de macromoléculas determinadas experimentalmente que puede consultarse a través de distintos puntos de acceso. Vamos a utilizar el gestionado por RCSB (Research Collaboratory for Structural Bioinformatics), cuya base de datos se denomina RCSB PDB.
En la página inicial se encuentra un campo para realizar búsquedas y el cuadro principal con distintos tabuladores (con opciones que aparecen en el menú superior).
Los apartados Welcome y Learn contienen información general, divulgativa y educativa. Los apartados Deposit y Analyze son utilizados por usuarios avanzados. Permiten insertar nuevas estructuras, estudiar su calidad o comparar distintas estructuras. En el apartado Search podemos navegar a través del archivo y localizar estructuras individuales o por tipos. Por último, en el apartado Visualize encontraremos herramientas sencillas para crear y observar modelos gráficos de estructuras.
A cada estructura se le asigna un código de 4 dígitos (PDB-ID). Para acceder a los datos de una estructura determinada introduce su PDB-ID en la herramienta de búsqueda. Si desconoces ese código para la estructura que te interesa, puedes buscarla utilizando el nombre de la molécula, su función, el nombre del autor de la estructura, etc.
Por ejemplo, una de las proteínas con más estructuras en la base de datos es la hemoglobina. La primera estructura de hemoglobina fue determinada por Max Perutz, galardonado premio Nobel de química (1962) junto a John Kendrew. Esta estructura inauguró en 1976, junto a otras 12, la primera versión del Protein Data Bank.El PDB-ID de esta estructura es 1MBN.
Si queremos comparar estructuras de hemoglobina humana en ausencia y en presencia de oxígeno, utilizando datos recientes publicados en un mismo trabajo por Tame en 2006, realizaríamos la búsqueda "haemoglobin Tame 2006, encontrando las estructuras 2DN2 (sin oxígeno), 2DN1 (oxi-hemoglobina) y 2DN3 (con CO unido).
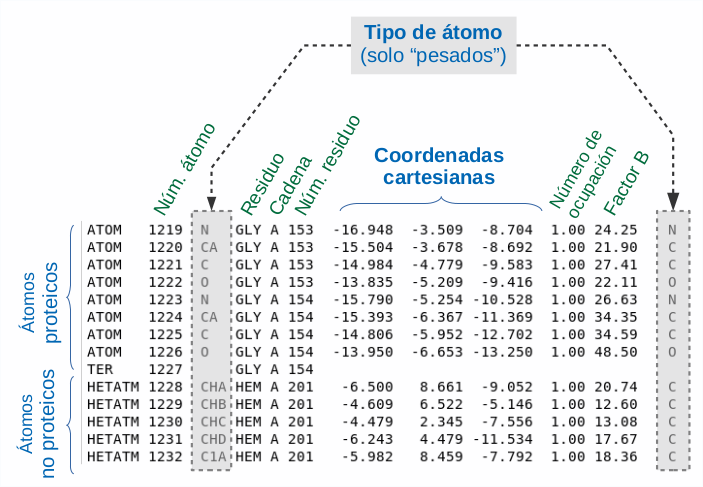
Información estructural en el formato PDB
Las estructuras de la base de datos se escriben en ficheros de texto con formatos especiales. Cuando accedemos a un registro, sus ficheros estructurales se encuentran en el apartado "Display Files".
El primero de los ficheros disponibles contiene solo la estructura primaria (secuencia de residuos), escrita en formato FASTA. Después se encuentran los ficheros de estructura tridimensional en distintos formatos. Entre ellos, el más intuitivo y utilizado recibe el nombre de formato "PDB".
En los ficheros PDB suele haber una primera parte, denominada HEADER con información complementaria de distinto tipo (sobre el tipo y origen de la molécula, el método usado para la determinación estructural, la calidad de la estructura, la secuencia de residuos, la estructura secundaria, etc).
La parte principal del fichero, en la que se define la estructura 3D, se encuentra a continuación y corresponde a todas las líneas que comienzan por ATOM. Si existen grupos químicos no proteicos en la molécula, su estructura se escribe en las lineas que comienzan por HETATM. En ambos casos, cada linea se organiza en columnas que contienen principalmente las denominaciones (tipo) de los átomos presentes en la estructura y sus coordenadas espaciales o cartesianas {x,y,z}. Por lo general se incluyen solo los átomos pesados (C, O, N, S, P u otros, si los hay, pero no H).
Como información adicional, en otras columnas aparecen también el número de orden de cada átomo, el nombre y orden de secuencia del residuo correspondiente y la denominación de la cadena a la que pertenece (con letra mayúscula). Por último, después de las coordenadas se encuentran dos columnas de números adicionales que corresponden al número de ocupación (frecuencia de esa posición espacial entre varias conformaciones posibles) y el factor "B" de temperatura (desplazamiento de la posición del átomo con respecto a un valor medio). Estos dos últimos números son una medida de la flexibilidad de la molécula en el punto correspondiente al átomo considerado.
Generalmente no existe información explícita sobre enlaces entre átomos en el fichero pdb. Sin embargo, dicha información no es necesaria para la reresentación de modelos moleculares de la estructura ya que los enlaces pueden definirse de manera automática por los programas informáticos de modelización teniendo en cuenta los tipos de átomo y las distancias entre ellos, calculadas a partir de su posición en el espacio.
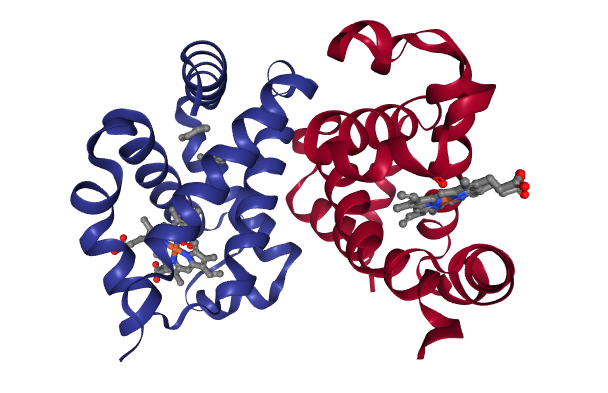
La unidad de asimetría de 2DN1 (oxi-hemoglobina) es un dímero αβ, distinto de la agrupación biológica esperada (tetrámero α2β2).
Dado que la mayoría de las estructuras de la base de datos han sido determinadas a partir de datos cristalográficos (mediante difracción de rayos X), generalmente, la estructura disponible corresponde a una unidad de asimetría, definida como la parte mas simple de la estructura cristalina que permite reconstruir una celda unidad (o unidad repetida del cristal). Sin embargo, la organización molecular funcional, llamada agrupación biológica (biological assembly) en muchos casos no coincide con la unidad de asimetría.
Un buen ejemplo de ello es el caso de estructuras de hemoglobina, para cual la agrupación biológica es un tetrámero (2x[αβ]), aunque en algunos casos la unidad de asimetría es dímerica (αβ).
Cuando la agrupación biológica y la unidad de asimetría no coinciden, la primera puede construirse a partir de la segunda utilizando la matriz de transformación proporcionada en el propio fichero PDB, en las lineas del HEADER marcadas con REMARK 350.
