Universitat de València - Máster en Aproximaciones Moleculares en Ciencias de la Salud
Tecnologías de la Medicina Molecular
Modelización y Análisis de Estructuras Macromoleculares
Utilizaremos el programa VMD (Visual Molecular Dynamics), desarrollado en la Universidad de Illinois y disponible en código abierto y de forma libre.
Para iniciarte en la utilización del programa puedes seguir esta Guía Básica de VMD. También puedes encontrar un manual de VMD en este enlace.
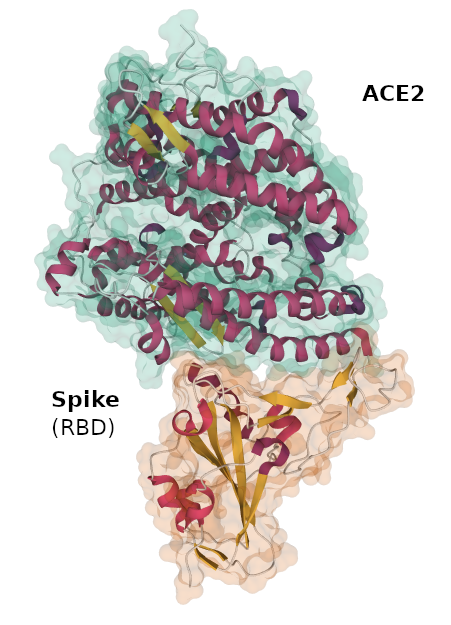
Complejo Spike-ACE2 (6LZG). La interacción de Spike tiene lugar prácticamente solo a través de la primera hélice.
Descarga el fichero de coordenadas pdb del dímero RBD-ACE2 (6M0J). Al cargar la estructura en VMD inicialmente obtienes una representación sencilla por defecto, que puedes cambiar según tus necesidades de análisis. Para conseguir una imagen más informativa, cambia la representación. Principalmente deberás utilizar las opciones que aparecen en Graphics -->Representations.
Primero llevaremos a cabo representaciones en distintas orientaciones en las que se destaquen los tipos de cadena y la disposición global de la molécula. El fichero pdb contiene coordenadas de un heterodimero, con las cadenas de proteína ACE2 (A ) y RBD de spike (E ). Contiene tambiém datos estructurales de moléculas no proteicas (múltiples moléculas de H2O, un ión Zn2+, varias moléculas del monosacárido NAG).
Crea representaciones separadas para cada una de las cadenas de proteína y para las moléculas no proteicas. Estudia con detenimiento las principales características de las cadenas de proteína (estructuras secundarias, propiedades de la superficie de cada cadena). Puedes moverte a lo largo de la cadena con ayuda de la herramienta Sequence viewer que encontrarás en Extensions→ Analysis. Para adaptar tu representación a lo que buscas combina opciones de DrawingMethod y ColoringMethod.
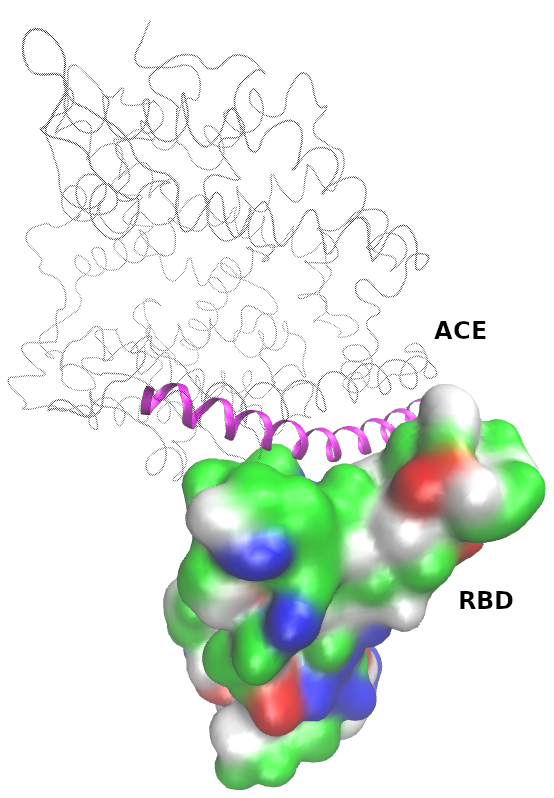
En representaciones separadas para cada cadena, elige superficie para una de ellas (DrawingMethod→ >QuickSurf), representando la otra de manera sencilla (DrawingMethod→ >tube). Colorea la representación de superficie según el tipo de residuo (ColoringMethod→ >ResType). Alterna este tipo de representación entre cadenas y observa qué tipo de residuos (polares, hidrofóbicos, con carga) se encuentran en la superficie de interacción.
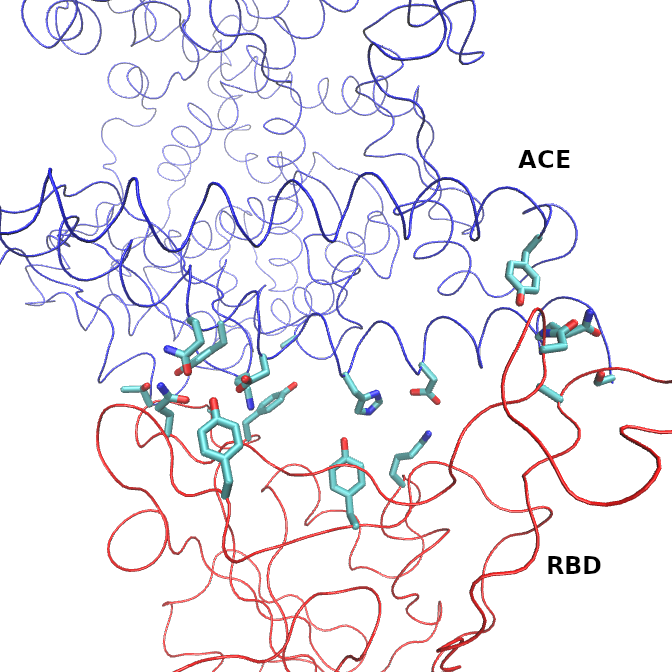
Residuos en la interfase de interacción entre Spike y ACE2 (6LZG)
Ahora buscaremos los residuos de una de las cadenas que se encuentran cercanos a residuos de la otra cadena.
Para ello, en una representación nueva, vamos a seleccionar los residuos de la cadena A con al menos un átomo que se encuentre al menos a 3 Å de cualquier átomo de la cadena E:
Para saber qué numeración corresponde a esos residuos, utiliza la opción Mouse→ Label→ Atoms (o bien teclea 1 con el puntero dentro del Display).
En una representación nueva, muestra los residuos de la cadena E cercanos a la cadena A.
Combina ambas representaciones con otras en las muestres las cadenas A y E de manera esquemática:
Para determinar la superficie de interacción entre ACE2 (A ) y RBD de spike (E ) utilizamos la consola Tk de VMD (Extensions -->Tk Console) y llevamos a cabo cálculos del area accesible al solvente (measure sasa). Lo hacemos de la manera siguiente:
Crea 'variables' independientes que contengan la selección de cadenas individuales "A" o "E", o el dímero "AE":
set chA [atomselect top "protein and chain A"]
set chE [atomselect top "protein and chain E"]
set chAE [atomselect top "protein and chain A E"]
Calcula las áreas accesibles para las cadenas individuales libres y en el dímero y para el dímero completo:
set areaA [measure sasa 1.4 $chA]
set areaE [measure sasa 1.4 $chE]
set areaAE [measure sasa 1.4 $chAE]
set areaA_AE [measure sasa 1.4 $chAE -restrict $chA]
set areaE_AE [measure sasa 1.4 $chAE -restrict $chE]
Calcula el área escondida en el dímero A:E debido a la interacción entre A y E
De la cadena A: expr ($areaA - $areaA_AE)
De la cadena E: expr ($areaE - $areaE_AE)
Del dímero completo: expr ($areaA + $areaE - $areaAE)
o también: expr ($areaA + $areaE - $areaA_AE - $areaE_AE)
Podemos restringir facilmente el cálculo de área accesible a un tipo determinado de residuo (hidrofóbico, polar, ácido, básico). Para ello solo tenemos que modificar las líneas de definición de las variables chA, chB, chAB, incluyendo en la selección el tipo de residuo ique queremos. Por ejemplo, set chAh [atomselect top "protein and chain A and hydrophobic"] crea la variable $chAh con los residuos hidrofóbicos de la cadena A y nos servirá para calcular el área de la superficie accesible restringida solo a los residuos hidrofóbicos de A.
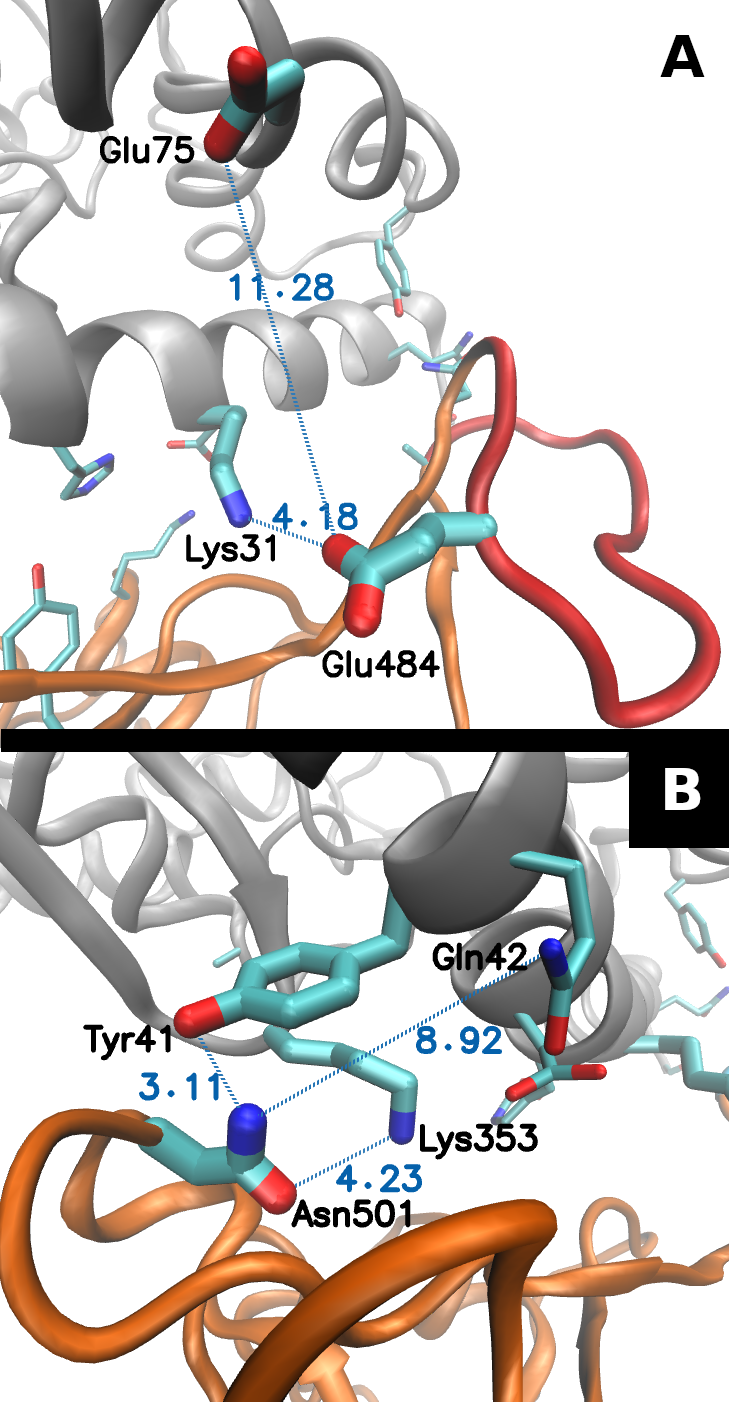
Detalle del entorno, en el complejo spike-ACE2 (6LZG), de residuos implicados en las mutaciones E484K (Glu484, A) y N501Y (Asn501, B) descritas recientemente en la proteína Spike del SARS-Cov-2.
Se han descrito muchas mutaciones en la proteína spike, algunas de las cuales aumentan la capacidad infectiva del virus. Entre los cambios mas estudiados se encuentran N501Y y E484K, denominados familiarmente mutaciones NellY y EriK, respectivamente. Es fácil localizar estos cambios en el contexto de la estructura del complejo spike-ACE e intentar entender su posible efecto.
Encontrar cada uno de los residuos implicados esas mutaciones es sencillo, si utilizamos su numeración de secuencia. Por ejemplo, para observar el residuo Glu484 (que cambia en la mutación EriK) creamos una representación con los siguientes parámetros:
Del mismo modo, el residuo Ans501, que cambia en la mutacón NellY, podemos representarlo como:
Cuando hemos representado ambos residuos podemos analizar su entorno e intentar entender si un cambio en las mutaciones señaladas podría afectar a las características de la interacción spike-ACE2.
Como vimos en la Parte 1, en la base de datos PDB existen estructuras experimentales de complejos RBD:ACE2 en los cuales la proteína vírica se encuentra mutada. Vamos a analizar la estructura de un caso sencillo (mutante Y453F, 7EKH) y compararlo con la estructura del dímero salvaje. Una manera sencilla de hacerlo es repetir el Apartado -2- para el dímero con el mutante Y453F de RBD, y comparar tus representaciones y áreas de superficie de interacción con los del dímero salvaje.
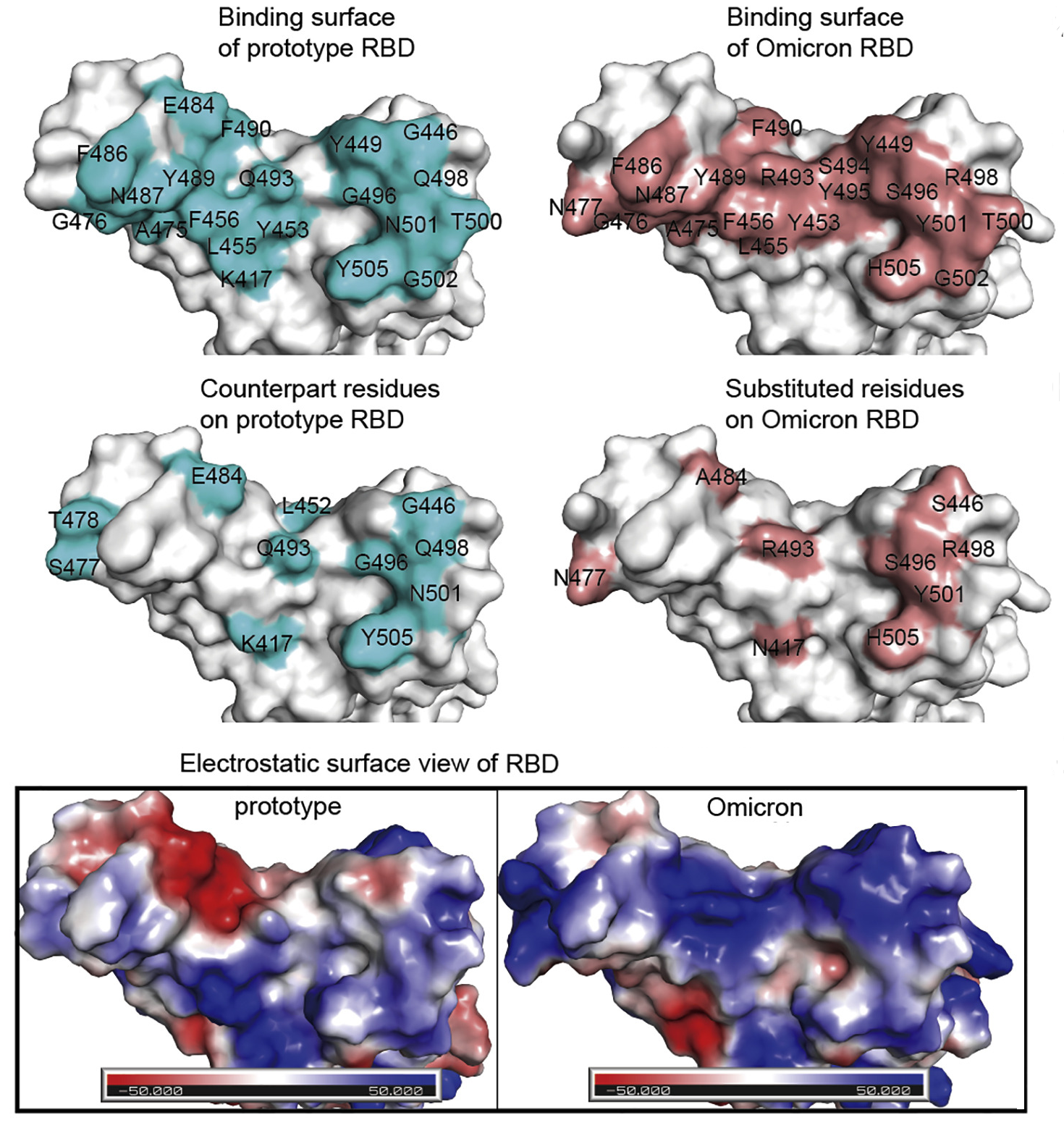
Superficie de interacción del dominio RBD de Spike con ACE2 en las variantes salvaje y Omicron del SARS-Cov-2 (7WBL, Han, et al. Cell 2022, in press).
La acumulación de mutaciones puede dar lugar a variantes del virus, algunas de las cuales son de especial interés por su aumento de la infectividad. En Noviembre de 2021 se describió la variante denominada Omicron, caracterizada por la presencia de un elevado número de mutaciones, algunas de las cuales afectan a residuos de la interacción entre el dominio RBD y ACE2.
Recientemente se han completado estudios estructurales del complejo RBD-ACE2 en varios mutantes del virus, incluida la variante Omicron (Han, et al. Cell 2022, in press, McCallum, et al. Science 2021, 0.1126/science.abn8652). Algunas de las estructuras correspondientes se encuentran accesibles en el PDB.
Es fácil comparar variantes del RBD de Spike en cuanto a su capacidad (estructural) de interacción con ACE2, en los casos wildtype y Omicron. Para el primero podemos utilizar el complejo que hemos usado en los apartados anteriores (6M0J). Para el segundo, busca en la base de datos PDB la estructura mas adecuada (ver Parte 1).
Para llevara a cabo la comparación, calcula, en cada caso, el área de las superficies del RBD y de ACE2 implicadas en la interacción entre ambas cadenas, y crea representaciones que te permitan observar diferencias entre las superficies y las interfaces de interacción.
