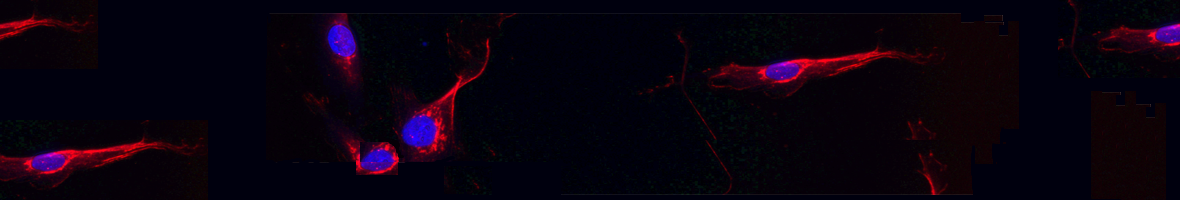
Myotonic Dystrophies
Myotonic dystyrophy (DM) encompassing DM1 and DM2, is caused by CTG and CCTG repeat expansions in the DMPK and CNBP genes, respectively. While both forms share common features like muscle wakness, myotonia, and multi-systemic symptoms, they differ in severity and onset. At our lab, we are dedicated to unconvering the mechanisms underlying muscle atrophy and gastrointestinal symptoms critical yet understudied aspects that significantly impac patients' quality of life. Our research involves developing innovatie preclinical models and pioneering oligonucleotide-based therapeutics. We are also at the forefront of collaborative initiatives, leading, the creation of the first Spanish registry for DM1 patients (the DM1-Hub project) and leveraging whole-genome sequencing to identify genetic modifiers. These efforts aim to deepen our undestanding of DM, improve patient care , and accelerate the development of effective therapies.
LGMDD2
Limb-girdle muscular dystrophy D2 (LGMDD2) is a genetic disorder caused by a 15-amino acid pathogenic tail in transportin-3 (TNPO3), which disrupts its nuclear import functions and impairs cellular homeostasis. Our lab has developed the first Drosophila, cell, and mouse models for LGMDD2, offering critical insights into the disease’s mechanisms and potential therapies. Currently, we are investigating subcellular proteomics and conducting collaborative structural studies to understand how this pathogenic tail alters TNPO3 function. Our efforts also focus on drug repurposing and using oligonucleotides to modulate TNPO3 expression to develop treatments for patients. Notably, LGMDD2 patients are naturally resistant to HIV infection due to TNPO3's crucial role in viral nuclear translocation.
Spinal Muscular Atrophy
Spinal muscular atrophy (SMA) is a genetic disorder characterized by motor neuron degeneration and progressive muscle weakness. Our lab has significant experience in identifying drugs that can be repurposed to treat SMA, accelerating the development of potential therapies. Building on this expertise, we are now focusing on the preclinical characterization of haloperidol, a promising candidate for SMA treatment. This includes unraveling its mechanism of action and exploring innovative delivery methods to optimize its efficacy and safety. This work is being carried out in close collaboration with leading SMA experts, ensuring rigorous evaluation and translational potential.
CANVAS
Cerebellar Ataxia, Neuropathy, and Vestibular Areflexia Syndrome (CANVAS) is a rare neurodegenerative disorder associated with progressive ataxia, sensory neuropathy, and vestibular dysfunction. This condition is frequently linked to an expanded AAGGG pentanucleotide repeat in the RFC1 gene, which disrupts normal cellular processes. Our team is developing the first Drosophila model to express this pathogenic repeat, leveraging the organism’s simplicity and genetic tractability for rapid preclinical studies. This model will enable the study of toxic DNA/RNA effects and facilitate drug screening for potential therapies. By combining molecular, cellular, and phenotypic analysis, this work aims to elucidate CANVAS mechanisms and identify therapeutic strategies, fostering innovation in rare disease research.
Oligonucleotide Therapeutics
We hace long focused on olignocleotide therapeutics, leveraging their precision to target genetic diseases. Previous successes include the development of antimiR strategies to modulate miRNA muscle regulators and gapmers, improving desease phenotypes in models of myotonic dystrophy. These studies underscored the importance of efficient delivery systems, as skeletal muscle uptake remains a major challenge. Building on this fundation, we are now advancing preclinical investiagions into optimized delivery methods, including bichromatic reporters to track intracelular oligonucleotide activity. Our focus extends to proteomic analyses to undestand cellular effects and structural studies of oligonucleotide-target interactions. These efforts aim to develop effective therapies, ensuring precise targeting and minimal toxicity, addressing unmet needs in muscle atrophy and related deseases.
Drosophilia as preclinical model
Our lab has extensive experience leveraging Drosophila as a robust preclinical model to study neuromuscular diseases and screen porential therapeutic compunds. These models are highly adventageous due to their rapid development cycles, genetic simplicity, and conserved molecular pathways relevant to human diseases. In previous studies, we developed and characterized Drosophila models for myotonic dystrophy, SMA and LGMDD2, successfuly using them to identify drug candidates. Currently, we are expanding this approach by generating precise trangenic models that mimic human pathogenic mechanisms. These models serve as platform for innovate therapeutic exploration, combining molecular phenotyping with high-trhoughput drug screening to accelerate translational research effors.