
-
Bioengineered in vitro 3D model of myotonic dystrophy type 1 human skeletal muscle
Fernández-Garibay X, Ortega MA, Cerro-Herreros E, Comelles J, Martínez E, Artero R, Fernández-Costa JM, Ramón-Azcón J - 2021 - Biofabrication
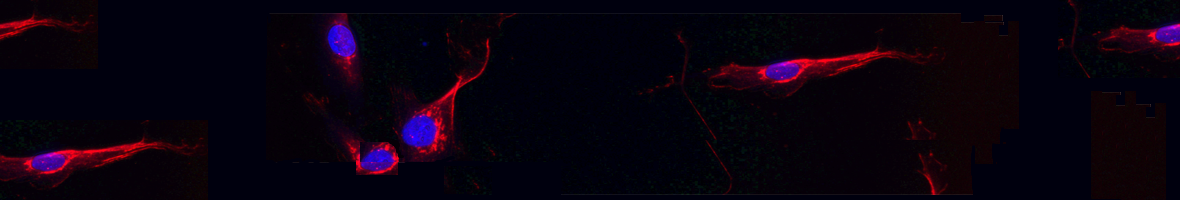
(2021).ArticleMyotonic dystrophy type 1 (DM1) is the most common hereditary myopathy in the adult population. The disease is characterized by progressive skeletal muscle degeneration that produces severe disability. At present, there is still no effective treatment for DM1 patients, but the breakthroughs in understanding the molecular pathogenic mechanisms in DM1 have allowed the testing of new therapeutic strategies. Animal models andin vitrotwo-dimensional cell cultures have been essential for these advances. However, serious concerns exist regarding how faithfully these models reproduce the biological complexity of the disease. Biofabrication tools can be applied to engineer human three-dimensional...
Myotonic dystrophy type 1 (DM1) is the most common hereditary myopathy in the adult population. The disease is characterized by progressive skeletal muscle degeneration that produces severe disability. At present, there is still no effective treatment for DM1 patients, but the breakthroughs in understanding the molecular pathogenic mechanisms in DM1 have allowed the testing of new therapeutic strategies. Animal models andin vitrotwo-dimensional cell cultures have been essential for these advances. However, serious concerns exist regarding how faithfully these models reproduce the biological complexity of the disease. Biofabrication tools can be applied to engineer human three-dimensional (3D) culture systems that complement current preclinical research models. Here, we describe the development of the firstin vitro3D model of DM1 human skeletal muscle. Transdifferentiated myoblasts from patient-derived fibroblasts were encapsulated in micromolded gelatin methacryloyl-carboxymethyl cellulose methacrylate hydrogels through photomold patterning on functionalized glass coverslips. These hydrogels present a microstructured topography that promotes myoblasts alignment and differentiation resulting in highly aligned myotubes from both healthy and DM1 cells in a long-lasting cell culture. The DM1 3D microtissues recapitulate the molecular alterations detected in patient biopsies. Importantly, fusion index analyses demonstrate that 3D micropatterning significantly improved DM1 cell differentiation into multinucleated myotubes compared to standard cell cultures. Moreover, the characterization of the 3D cultures of DM1 myotubes detects phenotypes as the reduced thickness of myotubes that can be used for drug testing. Finally, we evaluated the therapeutic effect of antagomiR-23b administration on bioengineered DM1 skeletal muscle microtissues. AntagomiR-23b treatment rescues both molecular DM1 hallmarks and structural phenotype, restoring myotube diameter to healthy control sizes. Overall, these new microtissues represent an improvement over conventional cell culture models and can be used as biomimetic platforms to establish preclinical studies for myotonic dystrophy.
Read moreHide DOI: 10.1088/1758-5090/abf6ae -
Rabphilin involvement in filtration and molecular uptake in Drosophila nephrocytes suggests a similar role in human podocytes
Selma-Soriano E, Llamusi B, Fernández-Costa JM, Ozimski LL, Artero R, Redón J - 2020 - Dis Model Mech
(2020).ArticleDrosophila nephrocytes share functional, structural and molecular similarities with human podocytes. It is known that podocytes express the rabphilin 3A (RPH3A)-RAB3A complex, and its expression is altered in mouse and human proteinuric disease. Furthermore, we previously identified a polymorphism that suggested a role for RPH3A protein in the development of urinary albumin excretion. As endocytosis and vesicle trafficking are fundamental pathways for nephrocytes, the objective of this study was to assess the role of the RPH3A orthologue in Drosophila, Rabphilin (Rph), in the structure and function of nephrocytes. We confirmed that Rph is required for the correct function of the endocytic...
Drosophila nephrocytes share functional, structural and molecular similarities with human podocytes. It is known that podocytes express the rabphilin 3A (RPH3A)-RAB3A complex, and its expression is altered in mouse and human proteinuric disease. Furthermore, we previously identified a polymorphism that suggested a role for RPH3A protein in the development of urinary albumin excretion. As endocytosis and vesicle trafficking are fundamental pathways for nephrocytes, the objective of this study was to assess the role of the RPH3A orthologue in Drosophila, Rabphilin (Rph), in the structure and function of nephrocytes. We confirmed that Rph is required for the correct function of the endocytic pathway in pericardial Drosophila nephrocytes. Knockdown of Rph reduced the expression of the cubilin and stick and stones genes, which encode proteins that are involved in protein uptake and filtration. We also found that reduced Rph expression resulted in a disappearance of the labyrinthine channel structure and a reduction in the number of endosomes, which ultimately leads to changes in the number and volume of nephrocytes. Finally, we demonstrated that the administration of retinoic acid to IR-Rph nephrocytes rescued some altered aspects, such as filtration and molecular uptake, as well as the maintenance of cell fate. According to our data, Rph is crucial for nephrocyte filtration and reabsorption, and it is required for the maintenance of the ultrastructure, integrity and differentiation of the nephrocyte.
Read moreHide DOI: 10.1242/dmm.041509 -
Therapeutic Potential of AntagomiR-23b for Treating Myotonic Dystrophy
Cerro-Herreros E, González-Martínez I, Moreno-Cervera N, Overby S, Pérez-Alonso M, Llamusí B, Artero R - 2020 - Mol Ther Nucleic Acids
(2020).ArticleMyotonic dystrophy type 1 (DM1) is a chronically debilitating, rare genetic disease that originates from an expansion of a noncoding CTG repeat in the dystrophia myotonica protein kinase (DMPK) gene. The expansion becomes pathogenic when DMPK transcripts contain 50 or more repetitions due to the sequestration of the muscleblind-like (MBNL) family of proteins. Depletion of MBNLs causes alterations in splicing patterns in transcripts that contribute to clinical symptoms such as myotonia and muscle weakness and wasting. We previously found that microRNA (miR)-23b directly regulates MBNL1 in DM1 myoblasts and mice and that antisense technology ("antagomiRs") blocking this microRNA (miRNA)...
Myotonic dystrophy type 1 (DM1) is a chronically debilitating, rare genetic disease that originates from an expansion of a noncoding CTG repeat in the dystrophia myotonica protein kinase (DMPK) gene. The expansion becomes pathogenic when DMPK transcripts contain 50 or more repetitions due to the sequestration of the muscleblind-like (MBNL) family of proteins. Depletion of MBNLs causes alterations in splicing patterns in transcripts that contribute to clinical symptoms such as myotonia and muscle weakness and wasting. We previously found that microRNA (miR)-23b directly regulates MBNL1 in DM1 myoblasts and mice and that antisense technology ("antagomiRs") blocking this microRNA (miRNA) boosts MBNL1 protein levels. Here, we show the therapeutic effect over time in response to administration of antagomiR-23b as a treatment in human skeletal actin long repeat (HSALR) mice. Subcutaneous administration of antagomiR-23b upregulated the expression of MBNL1 protein and rescued splicing alterations, grip strength, and myotonia in a dose-dependent manner with long-lasting effects. Additionally, the effects of the treatment on grip strength and myotonia were still slightly notable after 45 days. The pharmacokinetic data obtained provide further evidence that miR-23b could be a valid therapeutic target for DM1.
Read moreHide DOI: 10.1016/j.omtn.2020.07.021 -
Protective effects of mirtazapine in mice lacking the Mbnl2 gene in forebrain glutamatergic neurons: Relevance for myotonic dystrophy 1
Ramon-Duaso C, Rodríguez-Morató J, Selma-Soriano E, Fernández-Avilés C, Artero R, de la Torre R, Pozo ÓJ, Robledo P - 2020 - Neuropharmacology
(2020).ArticleMyotonic dystrophy type 1 (DM1) is a multisystemic disorder characterized by muscle weakness and wasting and by important central nervous system-related symptoms including impairments in executive functions, spatial abilities and increased anxiety and depression. The Mbnl2 gene has been implicated in several phenotypes consistent with DM1 neuropathology. In this study, we developed a tissue-specific knockout mouse model lacking the Mbnl2 gene in forebrain glutamatergic neurons to examine its specific contribution to the neurobiological perturbations related to DM1. We found that these mice exhibit long-term cognitive deficits and a depressive-like state associated with neuronal loss,...
Myotonic dystrophy type 1 (DM1) is a multisystemic disorder characterized by muscle weakness and wasting and by important central nervous system-related symptoms including impairments in executive functions, spatial abilities and increased anxiety and depression. The Mbnl2 gene has been implicated in several phenotypes consistent with DM1 neuropathology. In this study, we developed a tissue-specific knockout mouse model lacking the Mbnl2 gene in forebrain glutamatergic neurons to examine its specific contribution to the neurobiological perturbations related to DM1. We found that these mice exhibit long-term cognitive deficits and a depressive-like state associated with neuronal loss, increased microglia and decreased neurogenesis, specifically in the dentate gyrus (DG). Chronic treatment with the atypical antidepressant mirtazapine (3 and 10 mg/kg) for 21 days rescued these behavioral alterations, reduced inflammatory microglial overexpression, and reversed neuronal loss in the DG. We also show that mirtazapine re-established 5-HT1A and histaminergic H1 receptor gene expression in the hippocampus. Finally, metabolomics studies indicated that mirtazapine increased serotonin, noradrenaline, gamma-aminobutyric acid and adenosine production. These data suggest that loss of Mbnl2 gene in the glutamatergic neurons of hippocampus and cortex may underlie the most relevant DM1 neurobiological and behavioral features, and provide evidence that mirtazapine could be a novel potential candidate to alleviate these debilitating symptoms in DM1 patients.
Read moreHide DOI: 10.1016/j.neuropharm.2020.108030 -
miR-7 Restores Phenotypes in Myotonic Dystrophy Muscle Cells by Repressing Hyperactivated Autophagy
Sabater-Arcis M, Bargiela A, Furling D, Artero R - 2020 - Mol Ther Nucleic Acids
(2020).ArticleUnstable CTG expansions in the 3' UTR of the DMPK gene are responsible for myotonic dystrophy type 1 (DM1) condition. Muscle dysfunction is one of the main contributors to DM1 mortality and morbidity. Pathways by which mutant DMPK trigger muscle defects, however, are not fully understood. We previously reported that miR-7 was downregulated in a DM1 Drosophila model and in biopsies from patients. Here, using DM1 and normal muscle cells, we investigated whether miR-7 contributes to the muscle phenotype by studying the consequences of replenishing or blocking miR-7, respectively. Restoration of miR-7 with agomiR-7 was sufficient to rescue DM1 myoblast fusion defects and myotube growth....
Unstable CTG expansions in the 3' UTR of the DMPK gene are responsible for myotonic dystrophy type 1 (DM1) condition. Muscle dysfunction is one of the main contributors to DM1 mortality and morbidity. Pathways by which mutant DMPK trigger muscle defects, however, are not fully understood. We previously reported that miR-7 was downregulated in a DM1 Drosophila model and in biopsies from patients. Here, using DM1 and normal muscle cells, we investigated whether miR-7 contributes to the muscle phenotype by studying the consequences of replenishing or blocking miR-7, respectively. Restoration of miR-7 with agomiR-7 was sufficient to rescue DM1 myoblast fusion defects and myotube growth. Conversely, oligonucleotide-mediated blocking of miR-7 in normal myoblasts led to fusion and myotube growth defects. miR-7 was found to regulate autophagy and the ubiquitin-proteasome system in human muscle cells. Thus, low levels of miR-7 promoted both processes, and high levels of miR-7 repressed them. Furthermore, we uncovered that the mechanism by which miR-7 improves atrophy-related phenotypes is independent of MBNL1, thus suggesting that miR-7 acts downstream or in parallel to MBNL1. Collectively, these results highlight an unknown function for miR-7 in muscle dysfunction through autophagy- and atrophy-related pathways and support that restoration of miR-7 levels is a candidate therapeutic target for counteracting muscle dysfunction in DM1.
Read moreHide DOI: 10.1016/j.omtn.2019.11.012 -
Drosophila SMN2 minigene reporter model identifies moxifloxacin as a candidate therapy for SMA
Konieczny P, Artero R - 2020 - FASEB J
(2020).ArticleSpinal muscular atrophy is a rare and fatal neuromuscular disorder caused by the loss of alpha motor neurons. The affected individuals have mutated the ubiquitously expressed SMN1 gene resulting in the loss or reduction in the survival motor neuron (SMN) protein levels. However, an almost identical paralog exists in humans: SMN2. Pharmacological activation of SMN2 exon 7 inclusion by small molecules or modified antisense oligonucleotides is a valid approach to treat SMA. Here we describe an in vivo SMN2 minigene reporter system in Drosophila motor neurons that serves as a cost-effective, feasible, and stringent primary screening model for identifying chemicals capable of crossing the...
Spinal muscular atrophy is a rare and fatal neuromuscular disorder caused by the loss of alpha motor neurons. The affected individuals have mutated the ubiquitously expressed SMN1 gene resulting in the loss or reduction in the survival motor neuron (SMN) protein levels. However, an almost identical paralog exists in humans: SMN2. Pharmacological activation of SMN2 exon 7 inclusion by small molecules or modified antisense oligonucleotides is a valid approach to treat SMA. Here we describe an in vivo SMN2 minigene reporter system in Drosophila motor neurons that serves as a cost-effective, feasible, and stringent primary screening model for identifying chemicals capable of crossing the conserved Drosophila blood-brain barrier and modulating exon 7 inclusion. The model was used for the screening of 1100 drugs from the Prestwick Chemical Library, resulting in 2.45% hit rate. The most promising candidate drugs were validated in patient-derived fibroblasts where they proved to increase SMN protein levels. Among them, moxifloxacin modulated SMN2 splicing by promoting exon 7 inclusion. The recovery of SMN protein levels was confirmed by increased colocalization of nuclear gems with Cajal Bodies. Thus, a Drosophila-based drug screen allowed the discovery of an FDA-approved small molecule with the potential to become a novel therapy for SMA.
Read moreHide DOI: 10.1096/fj.201802554RRR -
Increased Muscleblind levels by chloroquine treatment improve myotonic dystrophy type 1 phenotypes in in vitro and in vivo models
Bargiela A, Sabater-Arcis M, Espinosa-Espinosa J, Zulaica M, Lopez de Munain A, Artero R - 2019 - Proc Natl Acad Sci U S A
(2019).ArticleMyotonic dystrophy type 1 (DM1) is a life-threatening and chronically debilitating neuromuscular disease caused by the expansion of a CTG trinucleotide repeat in the 3' UTR of the DMPK gene. The mutant RNA forms insoluble structures capable of sequestering RNA binding proteins of the Muscleblind-like (MBNL) family, which ultimately leads to phenotypes. In this work, we demonstrate that treatment with the antiautophagic drug chloroquine was sufficient to up-regulate MBNL1 and 2 proteins in Drosophila and mouse (HSALR) models and patient-derived myoblasts. Extra Muscleblind was functional at the molecular level and improved splicing events regulated by MBNLs in all disease models. In vivo,...
Myotonic dystrophy type 1 (DM1) is a life-threatening and chronically debilitating neuromuscular disease caused by the expansion of a CTG trinucleotide repeat in the 3' UTR of the DMPK gene. The mutant RNA forms insoluble structures capable of sequestering RNA binding proteins of the Muscleblind-like (MBNL) family, which ultimately leads to phenotypes. In this work, we demonstrate that treatment with the antiautophagic drug chloroquine was sufficient to up-regulate MBNL1 and 2 proteins in Drosophila and mouse (HSALR) models and patient-derived myoblasts. Extra Muscleblind was functional at the molecular level and improved splicing events regulated by MBNLs in all disease models. In vivo, chloroquine restored locomotion, rescued average cross-sectional muscle area, and extended median survival in DM1 flies. In HSALR mice, the drug restored muscular strength and histopathology signs and reduced the grade of myotonia. Taken together, these results offer a means to replenish critically low MBNL levels in DM1.
Read moreHide DOI: 10.1073/pnas.1820297116 -
MicroRNA-Based Therapeutic Perspectives in Myotonic Dystrophy
López Castel A, Overby SJ, Artero R - 2019 - Int J Mol Sci
(2019).ArticleMyotonic dystrophy involves two types of chronically debilitating rare neuromuscular diseases: type 1 (DM1) and type 2 (DM2). Both share similarities in molecular cause, clinical signs, and symptoms with DM2 patients usually displaying milder phenotypes. It is well documented that key clinical symptoms in DM are associated with a strong mis-regulation of RNA metabolism observed in patient's cells. This mis-regulation is triggered by two leading DM-linked events: the sequestration of Muscleblind-like proteins (MBNL) and the mis-regulation of the CUGBP RNA-Binding Protein Elav-Like Family Member 1 (CELF1) that cause significant alterations to their important functions in RNA processing. It...
Myotonic dystrophy involves two types of chronically debilitating rare neuromuscular diseases: type 1 (DM1) and type 2 (DM2). Both share similarities in molecular cause, clinical signs, and symptoms with DM2 patients usually displaying milder phenotypes. It is well documented that key clinical symptoms in DM are associated with a strong mis-regulation of RNA metabolism observed in patient's cells. This mis-regulation is triggered by two leading DM-linked events: the sequestration of Muscleblind-like proteins (MBNL) and the mis-regulation of the CUGBP RNA-Binding Protein Elav-Like Family Member 1 (CELF1) that cause significant alterations to their important functions in RNA processing. It has been suggested that DM1 may be treatable through endogenous modulation of the expression of MBNL and CELF1 proteins. In this study, we analyzed the recent identification of the involvement of microRNA (miRNA) molecules in DM and focus on the modulation of these miRNAs to therapeutically restore normal MBNL or CELF1 function. We also discuss additional prospective miRNA targets, the use of miRNAs as disease biomarkers, and additional promising miRNA-based and miRNA-targeting drug development strategies. This review provides a unifying overview of the dispersed data on miRNA available in the context of DM.
Read moreHide DOI: 10.3390/ijms20225600 -
Development of a Drosophila melanogaster spliceosensor system for in vivo high-throughput screening in myotonic dystrophy type 1
García-Alcover I, Colonques-Bellmunt J, Garijo R, Tormo JR, Artero R, Álvarez-Abril MC, López Castel A, Pérez-Alonso M
(2014).Article7:1297-1306
-
The use of whole-mount in situ hybridization to illustrate gene expression regulation
Llamusí B, Muñoz-Soriano V, Paricio N, Artero R
(2014).Article42:339-347
 University Institute of Biotechnology and Biomedicine (BIOTECMED)
University Institute of Biotechnology and Biomedicine (BIOTECMED)


© 2026 UV. - Av. Vicent Andrés Estellés, 19. 46100. Burjassot. Valencia. Spain. Tel. +34 963 543 460