.svg)




Specialists of the I2SysBio, CSIC and FISABIO analyse the genetic keys of tuberculosis virulence
- Scientific Culture and Innovation Unit
- June 19th, 2019
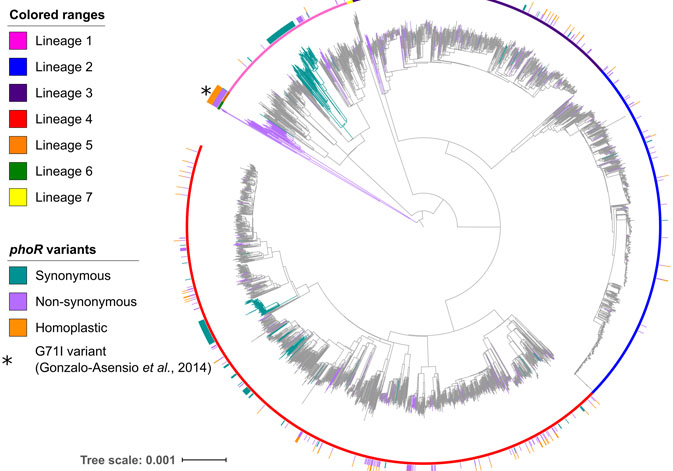
Phylogeny built with the genomes of more than 4,000 tuberculosis isolates from around the world. Those isolates that have accumulated mutations in the phoR gene are marked. / CSIC.
Research staff of the Institute of Biomedicine of Valencia (CSIC), the Institute for Integrative Systems Biology (CSIC-UV) and the Foundation for the Promotion of Health and Biomedical Research of the Valencian Government have analysed the genomic determinants of speciation and spread of tuberculosis. The work, published in the Science Advances journal, identifies the phoR gene as the key to a system involved in the virulence of tuberculosis and broadens the knowledge on the evolution of the bacteria that cause this pathology in animals and humans.
Tuberculosis is an infectious disease caused by the Mycobacterium tuberculosis bacterium, which causes a devastating mortality in humans and animals, and also leads to significant economic losses. Knowing how different bacterial lineages differ increases the understanding of the origins of the bacteria that causes the disease and the genetic mechanisms involved.
The researcher of the Institute of Biomedicine of Valencia (IBV, CSIC) Iñaki Comas and the researcher of the Mixed Unit FISABIO / Universitat de València-I2SysBio Álvaro Chiner (currently, member of the IBV, CSIC) explain that “to know the population genomic events that led to the birth of the tuberculosis pathogen, we have worked with the Mycobacterium tuberculosis Complex or MTBC, which includes a group of mycobacteria consisting of Mycobacterium tuberculosis and Mycobacterium africanum, which affect humans, as well as a series of pathogens isolated from other mammalian species known as Mycobacterium bovis, Mycobacterium pinnipedii, Mycobacterium orygis and Mycobacterium microti, among others”.
The increasing availability of population genomic data has allowed a better understanding of the genotypic and ecological differentiation between closely related bacteria. This has allowed the development of theoretical models of how the bacterial species and the genomic regions involved emerge.
“This study applies these models for the first time to a pathogen that affects humans, for which we have studied the bacteria most closely related to MTBC, known as Mycobacterium canettii or MCAN, an isolated strain of the Horn of Africa. Our analysis confirms the hypothesis that both shared a common gene pool. In addition, we have taken advantage of the availability of genomic sequences from thousands of clinical strains of the MTBC complex, as well as close relatives such as MCAN, to identify new genomic determinants in the appearance and subsequent MTBC spread”, Iñaki Comas adds.
Researchers have identified the phoR gene as the key to a system involved in virulence, which played a fundamental role in the evolution of the Mycobacterium tuberculosis complex. “Previous work had shown that phoR mutations played a central role in adapting the pathogen to different host species. We have shown the linkage of the phoR gene with the early spread of human tuberculosis, as well as in subsequent expansions. Our work also shows that the study of the evolution of pathogens helps to understand the determinants of their virulence past and present”, Comas concludes.
This work has also involved researchers from the CIBER of Epidemiology and Public Health of Valencia, the University of Oslo, the University of Helsinki, the Swiss Tropical and Public Health Institute of Switzerland, the University of Basel, the Microbiotica BioData Innovation Centre of the United Kingdom, and the Francis Crick Institute of the United Kingdom.
Article: Á. Chiner-Oms, L. Sánchez-Busó, J. Corander, S. Gagneux, S. Harris, D. Young, F. GonzálezCandelas, I. Comas. «Genomic determinants of speciation and spread of the Mycobacterium tuberculosis complex». Science Advances. DOI: 10.1126/sciadv.aaw3307



Tags
Covid
Culture
Studies
Institutional
Organisation
People
Research, innovation and transfer
University and Society
Living at the University